Clinical Summary
A 40-year-old man presents with a large, painless mass of the left thigh. The mass is resected following diagnostic needle biopsies. On gross examination, the mass measures 15 cm in maximum dimension and is relatively well-circumscribed with a multinodular appearance. The cut surface is glistening and gelatinous, without areas of necrosis.
Master List of Diagnoses
- Myxoid liposarcoma (MLS)
- Pleomorphic liposarcoma (PLS)
- Spindle cell lipoma
- Well-differentiated liposarcoma (WD-LPS)
Archive Case and Diagnosis
This case first appeared as Performance Improvement Program in Surgical Pathology (PIP) 2020, Case 02, and is myxoid liposarcoma of the thigh.
The information provided in this case was accurate and correct at the time of publication in 2020.
Any changes in terminology since the time of publication may not be reflected in this case.
Criteria for Diagnosis and Comments
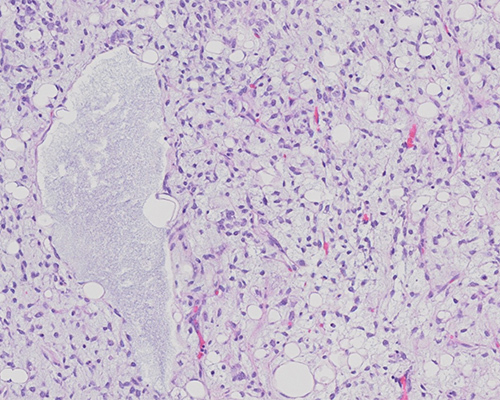
Histologic sections show a mixture of uniform round to oval-shaped cells admixed with signet ring lipoblasts in a prominent myxoid stroma. The stroma is further characterized by a delicate, branching capillary vasculature (“chicken-wire” pattern). Areas of mucin pools imparting a microcystic lymphangioma-like growth pattern are present. The neoplastic cells show minimal nuclear pleomorphism and lack prominent spindling or significant mitotic activity. These findings are consistent with myxoid liposarcoma (MLS).
MLS is the second most common type of liposarcoma, accounting for 20% to 30% of liposarcomas. It typically occurs in the fourth and fifth decades of life, with no gender predilection. MLS commonly presents as a large painless mass within the deep soft tissue of the extremities and only rarely arises in the retroperitoneum or subcutaneous tissue. On gross examination, these neoplasms are well-circumscribed, multinodular, tumors that show a prominent glistening, gelatinous cut surface in low-grade myxoid areas and a fleshy tan surface in higher-grade areas. Gross evidence of necrosis is uncommon.
Histologically, MLS displays a nodular growth pattern with enhanced cellularity at the periphery of the lobules. The tumor is composed of a mixture of uniform round to oval-shaped non-lipogenic cells and signet ring lipoblasts in a prominent myxoid stroma that is rich in delicate arborizing capillaries with a “chicken-wire” pattern. Frequently, the extracellular mucin forms large confluent pools, creating a microcystic lymphangioma-like or “pulmonary edema”-like growth pattern. Nuclear pleomorphism, giant tumor cells, spindling, and mitotic activity are typically not prominent. A subset of MLS that shows histologic progression to hypercellular/round cell morphology is associated with poorer prognosis. These higher-grade areas are characterized by solid sheets of round cells with high nuclear-to-cytoplasmic ratio and no intervening myxoid stroma. Myxoid and hypercellular/round cell liposarcomas represent a histologic continuum and are characterized by a recurrent translocation t(12;16)(q13;p11) that results in a FUS::DDIT3 gene fusion. The presence of the FUS::DDIT3 fusion is highly sensitive and specific for MLS and is seen in more than 95% of cases. Approximately 5% of myxoid liposarcomas harbor a t(12;22)(q13;q12) translocation, which fuses DDIT3 with EWSR1 on 22q12.
Wide-local surgical resection is the treatment of choice for MLS. Adjuvant radiotherapy has been shown to be effective in local control of MLS. Higher histologic grade, often defined as greater than 5% round cell component, presence of necrosis and TP53, and CDKN2A alterations are predictors of unfavorable outcome in localized MLS. Low-grade MLS have a metastatic risk of less than 10%, while one-third of patients with high-grade tumors develop distant metastasis, highlighting the need for adequate sampling and identifying any areas of transition to hypercellular/round cell morphology for accurate grading and prognostication. In contrast to most other sarcomas, MLS tends to spread to serosal membranes, the abdominal cavity, and distant soft tissues and bones, even in the absence of lung metastases. In a significant number of cases, patients present with synchronous or metachronous multifocal disease. The outcome for patients with multifocal MLS is poor regardless of its morphology.
Pleomorphic liposarcoma (PLS) is the rarest subtype of liposarcoma, accounting for about 5% of all liposarcomas and occurring most commonly in the extremities, while the trunk, retroperitoneum, and spermatic cord are less frequently affected. Histologically, PLS has infiltrative margins and contains a varying number of lipoblasts in a background of a high-grade pleomorphic sarcoma. The presence of lipoblasts is essential for diagnosis, highlighting the importance of adequate sampling. In most cases, the non-lipogenic component resembles undifferentiated pleomorphic sarcoma, with spindle and multinucleated giant cells, some of which are extremely large and often contain clear or vacuolated cytoplasm. PLS shows complex karyotypic abnormalities similar to other pleomorphic sarcomas and does not include the FUS::DDIT3 fusion.
Spindle cell lipoma typically occurs as an asymptomatic subcutaneous mass in the neck and upper trunk in older males. It is characterized by an admixture of mature fat and bland spindled cells surrounded by thick “rope-like” collagen, diffuse sheets of collagen, and/or a myxoid matrix. Strong CD34 immunoreactivity is a consistent feature of the spindle cell component. Some spindle cell lipomas, the so-called "low-fat" and "fat-free” variants, may have little to no adipocytic component. Spindle cell lipomas lack the typical, prominent plexiform vascular pattern of MLS.
Well‐differentiated liposarcoma (WD-LPS) most commonly involves the deep soft tissues of the limbs, especially the thigh, followed by the retroperitoneum, the paratesticular area and the mediastinum. The term "well-differentiated liposarcoma" is used almost exclusively for tumors arising in the retroperitoneum, mediastinum, and pelvis, while the term "atypical lipomatous tumor" is used for those arising in the extremities. Histologically, WD-LPSs are composed predominantly of mature adipocytes showing significant variation in cell size and at least focal nuclear atypia. Scattered hyperchromatic and multinucleated stromal cells, which are frequently more numerous in fibrous bands, and varying numbers of lipoblasts are often present. Cellular areas and myxoid stroma may be present. Nearly all WD-LPSs, including the dedifferentiated LPS, harbor the characteristic gene amplifications of MDM2 and CDK4 on chromosome 12, but lack the FUS::DDIT3 fusion typical of MLS.
Supplementary Questions
- Which of the following is most distinctive of myxoid liposarcoma (MLS)?
- CDK4 gene amplification
- FUS::DDIT3 gene fusion
- MDM2 gene amplification
- Rope-like collagen
- Thick-walled convoluted blood vessels
- Which of the following is true of MLS?
- Distant metastases to lung frequently occur before other sites.
- Prognosis is independent of histologic grade.
- Radiation is not indicated for local control.
- Round cell change results in a distinct change in the cytogenetic profile.
- Synchronous or metachronous multifocal disease is common.
- Which of the following is most typical of pleomorphic liposarcoma?
- Admixture of mature fat cells and bland spindle cells in a myxoid matrix
- CDK4 gene amplification on chromosome 12
- Uniform round to oval cells, signet ring lipoblasts and myxoid background with a rich capillary network
- Non-lipogenic component resembling undifferentiated pleomorphic sarcoma
- Well-circumscribed margins histologically
References
- Antonescu, C R et al. “Monoclonality of multifocal myxoid liposarcoma: confirmation by analysis of TLS-CHOP or EWS-CHOP rearrangements.” Clinical cancer research: an official journal of the American Association for Cancer Research vol. 6,7 (2000): 2788-93.
- Billings S, Folpe AL. Diagnostically challenging spindle cell lipomas: a report of 34 "low-fat" and "fat-free" variants. Am J Dermatopathol. 2007;29(5):437-442.
- Dei Tos AP. Liposarcomas: diagnostic pitfalls and new insights. Histopathology. 2014;64(1):38-52.
- Dürr HR, Rauh J, Baur-Melnyk A, et al. Myxoid liposarcoma: local relapse and metastatic pattern in 43 patients. BMC Cancer. 2018;18(1):304
- Ware PL, Snow AN, Gvalani M, Pettenati MJ, Qasem SA. MDM2 copy numbers in well-differentiated and dedifferentiated liposarcoma: characterizing progression to high-grade tumors. Am J Clin Pathol. 2014;141(3):334-341.
- WHO Classification of Tumours Editorial Board. WHO Classification of Tumours. Soft Tissue and Bone Tumours.: IARC Press; 2020. pp. 42-44.
Answer Key
- FUS::DDIT3 gene fusion (b)
- Synchronous or metachronous multifocal disease is common.(e)
- Non-lipogenic component resembling undifferentiated pleomorphic sarcoma (d)

