Clinical Summary
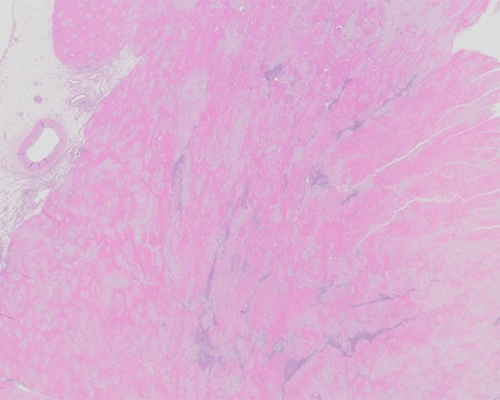
A 60-year-old man presents with dyspnea on exertion and swollen legs. Physical examination shows pitting edema in both legs and ascites. Electrocardiogram shows low voltage with both ventricular and atrial thickening on echocardiogram. A clinical history reveals symptoms of carpal tunnel syndrome for the preceding six months and a 10-pound weight loss with feelings of general malaise. An extensive laboratory evaluation reveals an M-spike with a monoclonal IgG kappa banding pattern, and bone marrow biopsy shows a kappa-restricted plasma cell myeloma. Induction chemotherapy is initiated, but the patient has a rapidly declining clinical course and expires. Autopsy examination reveals a 650-gram heart.
Master List of Diagnoses
- Cardiac amyloidosis
- Cardiac sarcoidosis
- Hypertrophic cardiomyopathy
- Loeffler endomyocardial fibrosis
- Myocardial infarction
Archive Case and Diagnosis
This case first appeared as Performance Improvement Program in Surgical Pathology (PIP) 2019, Case 31, and is cardiac amyloidosis (heart).
Criteria for Diagnosis and Comments
Microscopic sections of the heart at autopsy show diffuse atrial and ventricular extracellular deposition of amorphous proteinaceous material. Polarized Congo red microscopy shows apple-green birefringence, consistent with amyloid. In addition, myocarditis with increased numbers of eosinophils is noted in most of these tissue sections without evidence of granulomas. This eosinophilic myocarditis likely accounts for the rapid clinical decline and may be related to previous chemotherapy in this patient. However, the primary diagnosis is cardiac amyloidosis.
Amyloidosis is a rare group of diseases that are caused by the extracellular deposition of insoluble autologous proteins with an abnormal structure. Amyloid consists of approximately 95% misfolded protein fibrils in a β-pleated sheet configuration, with the remaining 5% of the protein structure comprised of chaperone “P” (or pentameric) components. More than 30 types of in vivo amyloid fibrils have been identified to date, causing either “systemic” or “localized” amyloidosis syndromes. Sequelae and clinical symptomatology are secondary to the physical accumulation of the abnormal amyloid protein within specific organs. Systemic amyloidosis has an estimated incidence of 4 in 1,000,000 and affects middle-aged men and women, with some variability based on specific amyloid subtypes. All cardiac amyloidoses, irrespective of the specific fibril protein, cause a restrictive cardiomyopathy leading to heart failure due to amyloid deposition. Cardiac amyloidosis is also frequently associated with peripheral neuropathy, carpal tunnel syndrome, and/or spinal stenosis, all of which can precede heart failure by months or even years.
In general, the gold-standard technique for diagnosis of amyloid is endomyocardial tissue biopsy with polarized Congo red histochemistry demonstrating apple-green birefringence. It is important to note that the typical pink-red staining of amorphous deposits on Congo red stain by brightfield examination is not diagnostic of amyloid, and it is only through polarized microscopy that the diagnosis is confirmed. Electron microscopy can also be used to confirm a diagnosis of amyloid. Other techniques, including immunostains and immunofluorescence, can be helpful but are less specific than diagnosis via Congo red.
The most common amyloid subtypes to affect the heart are systemic amyloidosis associated with clonal light chains (AL), transthyretin amyloidosis (ATTR), and isolated atrial amyloidosis (IAA) composed of atrial natriuretic peptide. The AL and ATTR types account for more than 95% of all cardiac amyloid disease. Amyloid A rarely affects the heart. Tests such as serum protein electrophoresis, immunofluorescence, immunohistochemistry, liquid chromatography/mass spectrometry, technetium scans, and cardiac magnetic resonance imaging can be helpful for characterization.
AL is a rare clonal plasma cell disease marked by the overproduction and misfolding of immunoglobulin light chain fragments. More than 3,000 new cases are diagnosed annually in the United States, with a median age at diagnosis of 63. It is a systemic disease and most often affects the heart, kidneys, and gastrointestinal tract. AL is an aggressive disease with median survival of 6 months from diagnosis if left untreated. In comparison, ATTR amyloidosis is due to acquired or inherited mutations in the transthyretin gene causing mutant transthyretin protein misfolding and amyloid deposition. The median age of onset is 75 years, with a male predominance. While ATTR amyloidosis portends a better prognosis than AL, it is still a progressive disease with median survival from diagnosis of 4 years or more, depending on the specific mutation causing the disease.
Cardiac sarcoidosis (CS) is a relatively rare, but increasingly recognized, manifestation of systemic sarcoidosis. CS is estimated to occur in 5% to 10% of all patients with systemic sarcoidosis but may be asymptomatic and, therefore, under-recognized. Microscopically, CS is similar to sarcoidosis in other organ systems, with focal non-caseating granulomas and scattered chronic inflammatory infiltrates. Clinically, CS can cause conduction abnormalities, arrhythmias, heart failure, and sudden death, and thus may be considered in the clinical differential with amyloidosis. Microscopically, amyloidosis and CS do not usually resemble each other; however, the myocarditis is a confounding factor in this case.
Hypertrophic cardiomyopathy (HCM) is defined by the presence of left ventricular wall thickening without chamber dilatation that cannot be explained by an abnormal heart loading condition. HCM is the most common inherited cardiac disease, affecting 1 in 500 adults, and causative mutations have been identified in numerous sarcomeric protein genes. The disease may be either familial or sporadic, with 60% of familial cases having an autosomal dominant pattern of inheritance. HCM usually causes outflow obstruction, angina, and palpitations and is usually not clinically confused with amyloidosis. Microscopically, HCM shows hypertrophic cardiac myocytes, disorganized myocardial architecture, and bizarre, branching myocyte forms with a whorled appearance. Myocytes are often at right angles to each other. Replacement myocardial fibrosis can appear amorphous, but the distribution and amount of fibrosis is usually less when compared to the amount of amorphous amyloid material in cardiac amyloidosis.
Loeffler endomyocardial fibrosis (LEMF) is a rare disease of unknown etiology in which the endocardium is progressively replaced by fibrosis with eventual heart failure and a poor prognosis. The fibrosis affects both ventricles asymmetrically but does not usually involve the atria. In addition, eosinophilia is often identified microscopically. This fibrosis typically causes a restrictive cardiomyopathy reminiscent of cardiac amyloidosis. LEMF can also manifest endomyocardial eosinophilic infiltrates, and it should be noted that this review case shows more of a full-thickness myocardial distribution of inflammation than in LEMF. Finally, amyloid deposition is not a characteristic of LEMF.
Myocardial infarction (MI) is defined as ischemic necrosis of the myocardium, and the fibrosis associated with older MI can form focal space-occupying areas within the myocardium. In comparison, however, amyloid usually affects the atria and the ventricles in a diffuse fashion. Histologic examination of cardiac amyloidosis shows expansion of the myocardium by the extracellular protein rather than myocyte destruction and contraction of the myocardium in MI. Finally, Congo red histochemistry will finalize the correct diagnosis, if necessary.
Supplementary Questions
- The majority of cardiac amyloidosis is caused by which proteins?
- Immunoglobulin light chains and atrial natriuretic factor
- Immunoglobulin light chains and pentameric protein
- Immunoglobulin light chains and transthyretin
- Transthyretin and albumin
- Transthyretin and pentameric protein
- The diagnosis of cardiac amyloidosis is confirmed on endomyocardial biopsy via which of the following?
- Congo red polarized microscopy with apple-green birefringence
- Immunofluorescence staining for amyloid
- Immunostain for the specific amyloid subtype
- Kappa and lambda light chain immunohistochemistry
- Salmon-pink amorphous material on Congo red light microscopy
- Which of the following statements about transthyretin amyloidosis (ATTR) is true?
- ATTR amyloidosis is an acquired disease without familial inheritance
- ATTR has a poor prognosis
- ATTR is associated with isolated left ventricular hypertrophy
- ATTR is due to mutation in the IGH gene
- ATTR protein is produced by the kidneys and brain
References
- Alam A, Thampi S, Saba SG, Jermyn R. Loeffler endocarditis: A unique presentation of right-sided heart failure due to eosinophil-induced endomyocardial fibrosis. Clin Med Insights Case Rep. 2017;10:1-4.
- Brambatti M, Matassini MV, Adler ED, Klingel K, Camici PG, Ammirati E. Eosinophilic myocarditis: characteristics, treatment, and outcomes. J Am Coll Cardiol. 2017;70(19):2363-2375.
- Donnelly JP, Hanna M. Cardiac Amyloidosis: An update on diagnosis and treatment. Cleve Clin J Med. 2017;84(12 Suppl 3):12-26.
- Fikrle M, Palacek T, Kuchynka P, et al. Cardiac amyloidosis: a comprehensive review. Cor et Vasa. 2013;55(1):E60-E75.
- Isaza N, Bolen M, Griffin B, Popovic Z. Ibrutinib-induced acute eosinophilic myocarditis mimicking infiltrative cardiomyopathy. J Am Coll Cardiol. 2019;73(9):2887.
- Kyriakou P, Mouselimis D, Tsarouchas A, et al. Diagnosis of cardiac amyloidosis: a systematic review on the role of imaging and biomarkers. BMC Cardiovasc Disord. 2018;18(1):221.
Author
Robert A. Schwartz, MD, FCAP
Surgical Pathology Committee
Eastern Connecticut Health Network and Waterbury Hospital
Manchester, CT
Answer Key
- Immunoglobulin light chains and transthyretin (c)
- Congo red polarized microscopy with apple-green birefringence (a)
- ATTR has a poor prognosis (b)

