Clinical Summary
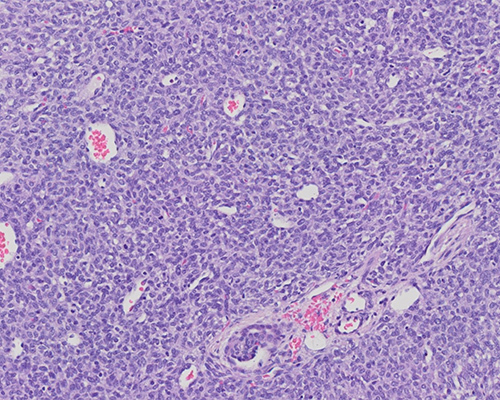
A 1-month-old boy presents with an abdominal mass. Imaging demonstrates a 10 cm solid mass centered in the right kidney. A nephrectomy is performed and shows an unencapsulated mass with tan cut surfaces and focal hemorrhage. The mass does not extend beyond the renal capsule. Immunohistochemical staining is positive for SMA and negative for desmin, WT1, and CD34. INI1 immunohistochemical expression is retained.
Master List of Diagnoses
- Clear cell sarcoma of the kidney
- Congenital mesoblastic nephroma
- Ewing sarcoma
- Nephroblastoma
- Rhabdoid tumor
Archive Case and Diagnosis
This case first appeared as Performance Improvement Program in Surgical Pathology (PIP) 2020, Case 11, and is congenital mesoblastic nephroma of the kidney. The information provided in this case was accurate and correct at the time of publication in 2020. Any changes in terminology since the time of publication may not be reflected in this case.
Criteria for Diagnosis and Comments
Histologic examination shows sheets of ovoid to spindle cells with eosinophilic cytoplasm, vesicular nuclei, small nucleoli, and frequent mitotic figures. Vessels have a hemangiopericytoma-like appearance. A few entrapped glomeruli and tubules are present within the tumor. The combined histologic and immunohistochemical features are consistent with congenital mesoblastic nephroma, cellular variant.
Congenital mesoblastic nephroma (CMN) is a mesenchymal tumor representing fewer than 5% of all pediatric renal tumors. It is the most common congenital renal neoplasm, with the majority of cases occurring in patients less than 1 year of age. Children typically present with an abdominal mass but may also present in utero with fetal hydrops and polyhydramnios. CMN is subclassified by histology: classic (25%), cellular (65%), and mixed (10%). Complete excision with wide margins is associated with an excellent prognosis, but tumor recurrence and metastases occur in 5% - 10% of patients. Risk factors associated with recurrence include cellular variant histology, age equal to or greater than 3 months, tumor rupture (Children’s Oncology Group System local stage III) and evidence of intrarenal or sinus lymphovascular invasion.
Grossly, CMN appears as an unencapsulated mass centered near the renal sinus with an indistinct border and bulging cut surface. Classic CMN typically has a firm cut surface with a whorled to trabeculated appearance. Cellular CMN more commonly demonstrates regions of necrosis, hemorrhage, and cysts.
Histologically, classic CMN is characterized by intersecting fascicles of spindle cells with tapered nuclei, eosinophilic cytoplasm, and rare mitoses. Background collagen deposition may be prominent. The border is indistinct and surrounds islands of normal renal parenchyma. Although not always present, necrosis and hemorrhage are common. Cellular CMN, morphologically identical to infantile fibrosarcoma, is densely cellular with more ovoid cells, poorly formed fascicles, sheet-like growth, a pushing border, and high mitotic activity. Regions of necrosis and hemorrhage are often present. Mixed CMN shows features of both classic and cellular CMN. By immunohistochemistry, tumor cells are positive for actin, pan-TRK and cyclin D1. INI1 expression is retained. There are no specific immunohistochemical stains to assist in diagnosis.
Molecular analysis demonstrates a recurrent chromosomal translocation t(12;15)(p13;q25) in cellular CMN. This translocation results in fusion of the ETV6 and NTRK3 genes and is identical to that seen in infantile fibrosarcoma. However, several variant fusions have been described, and absence of this translocation does not exclude the diagnosis of cellular CMN. Classic CMN is typically diploid and harbors an EGFR internal tandem duplication (ITD). EGFR ITD is more frequently identified in mixed CMN than the ETV6::NTRK3 gene fusion.
Clear cell sarcoma of the kidney (CCSK) is an aggressive renal neoplasm with a peak incidence at 2 years of age. The classic histology of CCSK is nests and cords of tumor cells within a network of fine fibrovascular septae. Many histologic patterns have been described, and these tumors may appear morphologically similar to many other pediatric renal neoplasms. CCSK is immunoreactive for cyclin D1 and BCOR and often positive for SATB2 and BCL2. Most tumors demonstrate internal tandem duplications of the BCOR gene. A minority have the chromosomal translocation t(10;17)(q22;p13) leading to a YWHAE::NUTM2 gene fusion.
Ewing sarcoma is rare in the kidney and typically occurs in young adults (average age: 20 years). The tumor is characterized histologically by sheets of small round blue cells with uniform nuclei and a small amount of clear cytoplasm. Immunohistochemistry shows diffuse membranous positivity for CD99 and nuclear positivity for NKX2.2. The majority of the tumors show t(11;22)(q24;q12), resulting in a EWSR1::FLI1 gene fusion, and a smaller number show t(21;22) with EWSR1::ERG gene fusion. Other fusion partners have been described.
Nephroblastoma (Wilms tumor) is the most common renal neoplasm in the pediatric population. The average age at diagnosis is 3 - 4 years of age. These tumors are commonly triphasic in appearance, with undifferentiated blastema, spindled stroma, and epithelial elements. The epithelial component may appear as tubules or glomeruli. The blastemal component is characterized by sheets of blastemal cells with minimal cytoplasm, frequent mitoses and small nucleoli. The blastemal and epithelial components are immunoreactive for WT1.
Rhabdoid tumor is an aggressive renal neoplasm with a poor prognosis. Most children are diagnosed prior to 2 years of age and present with advanced-stage disease. The tumor is composed of sheets of malignant cells with vesicular chromatin, prominent nucleoli and eosinophilic cytoplasm. The presence of cells with classic rhabdoid morphology is variable and may be sparse or focal. These tumors are associated with inactivation of the SMARCB1 tumor suppressor gene, leading to loss of INI1 expression by immunohistochemistry. A smaller subset of tumors demonstrates mutations in SMARCA4::BRG1.
Supplementary Questions
- Which of the following is true regarding congenital mesoblastic nephroma (CMN)?
- An INI1 immunohistochemical stain will show loss of expression.
- Average age of diagnosis is 5 years old.
- Classic CMN and infantile fibrosarcoma are morphologically identical and share the same recurrent genetic abnormality.
- Classic CMN shows formed fascicles of spindle cells and low mitotic activity.
- Prognosis is poor, with most patients developing recurrence and/or metastases within 5 years of nephrectomy.
- Which of the following is identified in cellular congenital mesoblastic nephroma?
- Internal tandem duplication in BCOR gene
- Mutation in SMARCB1
- t(10;17)YWHAE::NUTM2
- t(11;22) EWSR1::FLI1
- t(12;15) ETV6::NTRK3
- Which of the following increases the risk of recurrence of congenital mesoblastic nephroma?
- Cellular variant histology
- Complete excision with wide margins
- Diploid karyotype
- Presentation at 1 month of age
- Presentation in utero with polyhydramnios
References
- Argani P, Fritsch M, Kadkol SS, Schuster A, Beckwith JB, Perlman EJ. Detection of the ETV6-NTRK3 chimeric RNA of infantile fibrosarcoma/cellular congenital mesoblastic nephroma in paraffin-embedded tissue: application to challenging pediatric renal stromal tumors. Mod Pathol. 2000;13(1):29-36.
- Argani P, Calio A, Chang KTE, Cunha IW, de Ktijer RR, Vujanic GM. Congenital mesoblastic nephroma. In: WHO Classification of Tumours Editorial Board. Urinary and male genital tumours. International Agency for Research on Cancer; 2022. pp112 & 113
- Church AJ, Calicchio ML, Nardi V, et al. Recurrent EML4-NTRK3 fusions in infantile fibrosarcoma and congenital nephroma suggest a revised testing strategy. Mod Pathol. 2018;31(3):463-473.
- El Demellawy D, Cundiff CA, Nasr A, et al. Congenital mesoblastic nephroma: a study of 19 cases using immunohistochemistry and ETV6-NTRK3 fusion gene rearrangement. Pathology. 2016;48(1):47-50.
- Glick RD, Hicks MJ, Nuchtern JG, Wesson DE, Olutoye OO, Cass DL. Renal tumors in infants less than 6 months of age. J Pediatr Surg. 2004;39(4):522-525.
- Husain AN, Stocker JT, Dehner LP, eds. Stocker and Dehner’s Pediatric Pathology. 4th ed. Wolters Kluwer; 2016: 800-864.
- Sandberg AA, Bridge JA. Updates on the cytogenetics and molecular genetics of bone and soft tissue tumors: congenital (infantile) fibrosarcoma and mesoblastic nephroma. Cancer Genet Cytogenet. 2002;132(1):1-13.
Answer Key
- Classic CMN shows formed fascicles of spindle cells and low mitotic activity (d)
- t(12;15) ETV6::NTRK3 (e)
- Cellular variant histology (a)

