- Home
- Member Resources
- Pathology Case Challenge
- Retroperitoneum
Clinical Summary
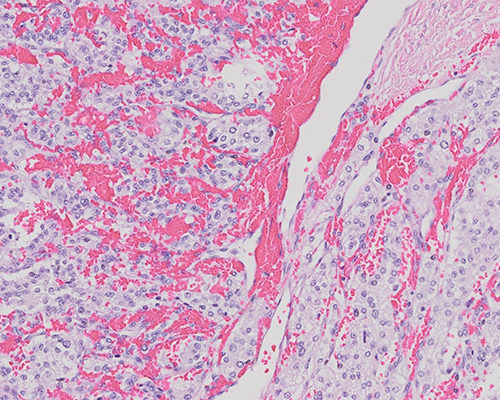
A 48-year-old man with a past medical history of hypertension, diabetes, and peripheral vascular disease presents with longstanding abdominal pain and distension. A computed tomography scan demonstrates a 12 cm midline retroperitoneal tumor. The excised tumor is a 320-gram, encapsulated, and lobulated mass measuring 12 cm in the largest dimension. Sectioning reveals a red-brown, focally myxoid cut surface with a yellow to orange rind. Tumor cells are negative for cytokeratins but positive for neuroendocrine markers (synaptophysin and chromogranin).
Master List of Diagnoses
- Adrenal cortical adenoma
- Alveolar soft part sarcoma
- Clear cell renal cell carcinoma
- Extra-adrenal paraganglioma (sympathetic paraganglioma)
- Well-differentiated neuroendocrine tumor
Archive Case and Diagnosis
This case first appeared as Performance Improvement Program in Surgical Pathology (PIP) 2020, Case 16, and is an extra-adrenal (sympathetic) paraganglioma. The information provided in this case was accurate and correct at the time of publication in 2020. Any changes in terminology since the time of publication may not be reflected in this case.
Criteria for Diagnosis and Comments
Sections show a partially encapsulated neoplasm composed of tumor cells arranged in alveolar and trabecular structures. Tumor cells are predominantly monomorphic in appearance with clear cytoplasm; the nuclei vary from small to somewhat large, with occasional nuclear pseudoinclusions. An occasional mitotic figure is identified. The tumor is highly vascularized and shows a network of capillaries rimming the nests of neoplastic cells. The central portion of the tumor shows hyalinization and hemosiderin-laden macrophages. Tumor cells are diffusely positive for vimentin, synaptophysin, and chromogranin and negative for cytokeratins. S100 highlights the intervening sustentacular cells. The histologic appearance and location within the retroperitoneum are consistent with a diagnosis of extra-adrenal paraganglioma (sympathetic paraganglioma).
Paragangliomas are most commonly seen in the 4th and 5th decade, with an equal sex predilection. They are non-epithelial, neuroendocrine neoplasms arising from the neural crest-derived paraganglion cells. The paraganglion system comprises the adrenal medulla and the extra-adrenal paraganglion system (which in turn can be either sympathetic or parasympathetic). The vast majority of paragangliomas arising from the sympathetic nervous system are located within the abdomen or pelvis (along the vertebral column), while the majority of parasympathetic paragangliomas are found in the head and neck region. Specifically, paragangliomas have been found to occur in the carotid body, jugulotympanic paraganglia, paracaval retroperitoneal sites and the organ of Zuckerkandl located at the bifurcation of the aorta or inferior mesenteric artery. Head and neck paragangliomas have several clinical synonyms based on the location of the tumor (eg, glomus jugulare tumor, carotid body tumor) and tend to be non-functional. On the other hand, extra-adrenal paragangliomas, also known as sympathetic paragangliomas, are more likely to be functional due to excess catecholamine (dopamine and norepinephrine) production. Paragangliomas of the adrenal medulla are specifically referred to as pheochromocytomas.
Paragangliomas are characteristically composed of small nests of chromaffin cells or chief cells and peripherally placed spindle cells, arranged in an organoid pattern, resulting in the so called zellballen appearance (German for “ball of cells”). Nuclear pleomorphism and nuclear pseudoinclusions are present, but mitotic activity is sparse. Other described morphological features include intracellular hyaline globules, intracytoplasmic melanin-like pigment deposition, clear cells, and oncocytic changes.
The tumor cells express the neuroendocrine markers synaptophysin, chromogranin, and CD56. They are negative for cytokeratin, SMA, desmin, and TFE3. The supporting sustentacular cells, when present, stain for S100 and SOX10. Sympathetic paragangliomas are more likely to be positive for chromogranin when compared to parasympathetic paragangliomas. Demonstration of dopamine beta-hydroxylase and tyrosine hydroxylase (catecholamine-synthesizing enzymes) may help in differentiating paragangliomas from other neuroendocrine tumors in the region.
While the majority of paragangliomas occur sporadically, at least 30% to 40% of the cases have been associated with germline mutations and/or hereditary settings. Paragangliomas are associated with several hereditary conditions, such as multiple endocrine neoplasia (MEN) types 2A and 2B (RET mutation), neurofibromatosis type 1 (von Recklinghausen disease, NF1 mutation), von Hippel–Lindau disease (VHL mutation), and Carney–Stratakis syndrome (succinate dehydrogenase [SDH] gene mutation). Germline mutations in the autosomal genes encoding SDH enzymes, including subunits A, B, C, D, and succinate dehydrogenase complex assembly factor 2 (SDHAF2), account for approximately 50% of the germline mutations in paragangliomas and pheochromocytomas. There seems to be some correlation between morphology and genetic alterations; for example, myxoid foci are more likely to be seen in tumors associated with VHL abnormalities, and tight nests of rounded clear epithelioid cells are more likely to be seen in SDH gene-mutated tumors. These morphological changes are merely clues and do not supplant the need to perform molecular/ancillary testing in these tumors.
The World Health Organization in 2004 defined malignancy in paragangliomas/pheochromocytomas as the presence of metastasis into a site where normal chromaffin cells are not present (eg, bone, lung, liver, lymph node). By current paradigms, all pheochromocytomas and paragangliomas are considered to have some metastatic potential. The risk for metastasis is higher in a sympathetic paraganglioma than parasympathetic paraganglioma or pheochromocytoma. The Pheochromocytoma of the Adrenal Gland Scaled Score (PASS) cannot be used to assess metastatic risk in extra-adrenal paragangliomas. Many histologic and immunophenotypic features such as tissue invasion, increased vascularization, and high Ki67 proliferation fraction have been proposed to predict malignancy but have not been found to be predictive of metastasis. Tumors greater than 5.0 cm, presence of SDHB mutation or loss of SDHB expression by immunohistochemistry and MAX mutations are associated with higher metastatic potential. SDHB loss by immunohistochemistry is a good surrogate marker and screening tool for triaging patients for further germline or somatic mutation analyses and genetic counseling. Stromal cells, endothelial cells, and inflammatory cells serve as good internal positive controls for SDHB staining. True immunoreactivity for SDHB imparts a strong, granular cytoplasmic staining, consistent with its mitochondrial localization. Complete absence of staining is commonly seen with SDHA, SDHB, and SDHC mutations, whereas SDHD mutations typically impart a nonspecific cytoplasmic blush and may be falsely interpreted as positive (ie, retained) expression.
As the name implies, adrenal cortical adenoma is a benign tumor of the adrenal cortex. The tumor cells may consist of a mix of lipid-rich, clear, vacuolated cells, and lipid-poor eosinophilic cells with compact cytoplasm. The neoplastic cells demonstrate positivity for adrenal cortical markers inhibin and MelanA. They are occasionally positive for synaptophysin but are typically negative for chromogranin. These tumors can also be associated with several hereditary conditions such as MEN type 1, McCune–Albright syndrome, Carney complex, Beckwith–Wiedemann syndrome, and congenital adrenal hyperplasia, among others.
Alveolar soft part sarcoma is a slow-growing malignant neoplasm characterized by a nested architecture with a pseudoalveolar growth pattern. This tumor is most frequently identified in infants, children, and young adults with a slight female predilection. Alveolar soft part sarcoma typically shows strong nuclear TFE3 positivity and is negative for synaptophysin, chromogranin, and S100. All cases of alveolar soft part sarcoma have been found to harbor the specific chromosomal translocation t(X;17)(p11;q25), which results in the ASPSCR1-TFE3 fusion product. Overall, despite the slow-growing nature of this tumor, prognosis is poor, with frequent late metastases.
Clear cell renal cell carcinoma is a malignant tumor that is usually limited to the renal parenchyma. The tumor frequently exhibits a golden-yellow gross appearance and abundant, clear cytoplasm due to the presence of intracytoplasmic lipid. The tumor cells are arranged in nests and are typically surrounded by abundant fibrovascular septations. Clear cell renal cell carcinoma is usually positive for CD10, PAX8, RCC, and carbonic anhydrase IX. The majority of these neoplasms are associated with mutations in the VHL gene or loss of chromosome 3p.
Well-differentiated neuroendocrine tumors are a heterogenous group of tumors with diverse clinical presentations depending on the organ of origin. Neuroendocrine tumors are typically positive for the neuroendocrine markers synaptophysin, chromogranin, and CD56. Diffuse positivity for cytokeratins helps distinguish neuroendocrine tumors from paragangliomas. Similar to paragangliomas, neuroendocrine tumors can be associated with several genetic syndromes, such as multiple endocrine neoplasia type 1 and von Hippel–Lindau syndrome.
Supplementary Questions
- Which of the following features is the best predictor of metastasis in cases of paraganglioma?
- Activating RET mutation
- Germline SDHB mutation
- Location within adrenal gland
- Nuclear atypia
- Vascular invasion
- Which of the following is true regarding SDHB immunohistochemistry?
- Cytoplasmic blush is indicative of retained expression of SDHB.
- Membranous staining is considered to be retained expression of SDHB.
- Nuclear staining is considered to be retained expression of SDHB.
- SDHB immunostaining is a poor screening test for evaluation of genetic alterations in the SDHB gene.
- Strong, granular cytoplasmic staining is considered to be retained expression of SDHB.
- Which of the following tumors demonstrates a nested histologic appearance with large eosinophilic cells, strong nuclear positivity for TFE3, and translocation t(X;17)?
- Alveolar soft part sarcoma
- Clear cell sarcoma
- Malignant melanoma
- Paraganglioma
- Translocation-associated renal cell carcinoma
References
- Asa S, Ezzat S, Mete O. The Diagnosis and Clinical Significance of Paragangliomas in Unusual Locations. J Clin Med. 2018;7(9):E280.
- Fliedner SM, Lehnert H, Pacak K. Metastatic Paraganglioma. Semin Oncol. 2010;37(6):627-637.
- Gill AJ. Succinate dehydrogenase (SDH)-deficient neoplasia. Histopathology. 2018;72(1):106-116.
- Giubellino A, Lara K, Martucci V et al. Urinary Bladder Paragangliomas: How Immunohistochemistry Can Assist to Identify Patients With SDHB Germline and Somatic Mutations. Am J Surg Pathol. 2015;39(11):1488-1492.
- Lloyd RV, Osamura RY, Kloppel G, Rosai J. WHO Classification of Tumours of Endocrine Organs. 4th ed. IARC Press; 2017.
- O’Riordain DS, Young WF Jr, Grant CS, Carney JA, van Heerden JA. Clinical Spectrum and Outcome of Functional Extraadrenal Paraganglioma. World J Surg. 1996;20(7):916-922.
- Tischler AS. Pheochromocytoma and Extra-adrenal Paraganglioma: Updates. Arch Pathol Lab Med. 2008;132(8):1272-1284.
- Thompson LD. Pheochromocytoma of the Adrenal gland Scaled Score (PASS) to separate benign from malignant neoplasms: a clinicopathologic and immunophenotypic study of 100 cases. Am J Surg Pathol. 2002;26(5):551-566.
- Turchini J, Cheung VKY, Tischler AS, De Krijger RR, Gill AJ. Pathology and genetics of phaeochromocytoma and paraganglioma. Histopathology. 2017;72(1):97-105.
Answer Key
- Germline SDHB mutation (b)
- Strong, granular cytoplasmic staining is considered to be retained expression of SDHB. (e)
- Alveolar soft part sarcoma (a)

