Below are answers to commonly asked questions pertaining to performance and interpretation of CAP PT/EQA programs. To make it easy to find answers, the questions are categorized by discipline.
General
The evaluation report lists your results, the statistics for your peer group, and your normalized results as a standard deviation index (SDI). This value is obtained by subtracting the peer group mean from your result and then dividing by the standard deviation.
The SDI is calculated from the unrounded (or more precise) figures. For small numbers, the differences attributed to rounding can be rather significant. Nevertheless, the “true” SDI is displayed on the evaluation report. The participant is not able to calculate SDI separately with the other rounded numbers provided on the evaluation report.
The CAP sometimes includes proficiency testing/external quality assessment (PT/EQA) specimens that assess the ability of laboratory staff to make difficult distinctions, to deal with special interferences or circumstances, or that may challenge the routine capabilities of many well-run laboratories. In these cases, the PT/EQA specimen is not graded by design. In some situations, an entire PT/EQA program may consist of educational, ungraded specimens designed to help subscribers improve their skills and capabilities. Educational PT/EQA challenges are designated with an evaluation code 26. These challenges are not formally graded, and laboratories should utilize data in the participant summery (PS) report to perform a self-evaluation. Common methods used are using alternative peer groups for quantitative data and surmising the correct response from other participants or PT/EQA providers intent for qualitative data.
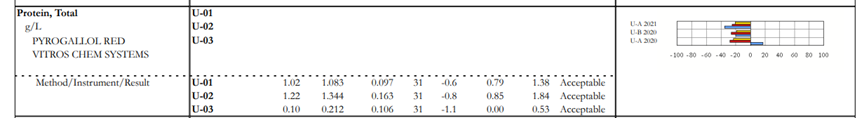

This happens when we have an alert on the result form. In some instances, a particular manufacturer has 2 different reagent lots in use. The manufacturer completed the studies and determined that these reagents performed differently on PT/EQA material. The CAP collects the data and forms peer group by method/instrument/reagent not to penalize laboratories using different reagents. When the laboratory selects a reagent (original or new) on the result form, the data will be displayed on top of the dotted line and peer group will be formed by method/instrument/reagent. When a laboratory does not select a reagent because it doesn’t apply to them, their data will be displayed on the bottom of the dotted line and the peer group will be formed by method/instrument.
Sometimes, fewer than 80% of participants or referees (for Clinical Laboratory Improvement Amendments of 1988 [CLIA] regulated analytes) agree on what is the correct response for a challenge. In this case, the challenge is not graded. The CAP does not believe it is reasonable to hold laboratories accountable for interpreting challenges that did not meet the expected criteria.
The minimum and maximum values provided in the PS report are not the limits of acceptability. These are the lowest and highest values reported for that peer group. The acceptable limits are located on your evaluation report.
Clerical errors cannot be regraded. Document that your laboratory performed a self-evaluation and compared its results to the intended response when provided in the PS report. Clerical errors may indicate a need for additional staff training, review of instructions provided with the PT/EQA program, addition of a second reviewer, or investigation of the reporting format provided by the testing device.
At minimum, any concern about the performance of any assay in the laboratory should trigger an informal process improvement assessment. A single unacceptable response (due to a clerical error) may not lead to significant change in the laboratory, but the cause of an unacceptable response must be determined, to the extent possible, and triaged appropriately by laboratory leadership. Conversely, investigation of a single unacceptable response could identify a situation requiring a complex improvement plan requiring assay re-validation. Therefore, review and assessment of all unacceptable responses, regardless of whether the laboratory achieves an overall acceptable score for the program, is recommended. An unsuccessful event indicates the laboratory did not achieve overall acceptable concordance with the intended responses (eg, did not achieve a passing score). In this situation, a comprehensive process improvement assessment should be initiated with appropriate corrective action taken for each unacceptable result.
ACCURACY
Accuracy-based specimens are collected by drawing donors and pooling the serum from multiple donors as described in the modified CLSI C37A Preparation and Validation of Commutable Frozen Human Serum Pools as Secondary Reference Materials for Cholesterol Measurement Procedures, Approved Guideline. There are no additives, preservatives, or spikes added to the specimens. These specimens should behave in the same manner as patient specimens.
BIOCHEMICAL AND MOLECULAR GENETICS
Here are the CAP recommendations and requirements for PT/EQA.
If you need further assistance, contact the Customer Contact Center and provide as much information as possible to help triage your request (eg, is your laboratory doing next-generation sequencing for somatic or germline purposes? Is your laboratory doing a panel, genome or exome testing? What genes is your laboratory testing for?).
Usually there is an extra space or inappropriate punctuation added to the customer’s result that might be difficult to see on the evaluation reports. Review the PS report for clarification. Laboratories should conform to the most recent HGVS recommendations. The following are inappropriate responses and are considered unacceptable:
- The use of “X” to indicate a translation stop codon
- Extra spaces (eg, c.145 G>A or p. Arg541*)
- Incorrect usage/mixing of upper and/or lowercase letters (eg, p.GLN541*, c.123c>t, C.Pro543Arg,p.Gln451ter)
- Reporting of the amino acid change inside parentheses (p.Gln541*). Per HGVS, the parentheses should be after the p., ie, p.(Gln541*)
- Incorrect short form for frame shifts (eg, p.Arg123Profs, p.Arg123fs*3). The short form should indicate the first amino acid changed, its position and “fs” without further detail (eg, p.Arg123fs)
- Missing punctuation (eg, c145G>A)
CHEMISTRY
The CAP PT/EQA is designed to assess how well laboratories perform direct analyte measurements. Most calculated analytes values except for hemoglobin (estimated), because it is a regulated analyte, should not be reported. The following are examples of analytes derived from a calculation that should not be reported: LDL cholesterol, total iron-binding capacity (TIBC), osmolality, and free testosterone.
The CAP PT/EQA is designed to assess how well laboratories perform direct analyte measurements. Oxygen saturation (calculated) is derived from a calculation using other directly measured blood gas analytes. The Blood Oximetry (SO) program includes reporting options for oxyhemoglobin but does not include a reporting option for oxygen saturation.
The 4th Universal Definition of Myocardial Infarction, along with the International Federation for Clinical Chemistry Committee on Clinical Applications of Cardiac Biomarkers (IFCC C-CB), recommend reporting high-sensitivity troponin concentrations in nanograms per liter (ng/L), as well as reporting results in whole numbers. Contemporary troponin results can be reported in micrograms per liter (μg/L) or nanograms per milliliter (ng/mL). In the current era of electronic medical records reporting, test results to multiple decimal places can lead to misinterpretation of results and contribute to medical error. Reporting high-sensitivity troponin results in whole numbers and with appropriate concentration units (ng/L) avoids ambiguity of reporting results containing numerous zeros after the decimal point.
CYTOPATHOLOGY
All individuals (physicians and cytotechnologists) who examine or interpret gynecologic cytology specimens (Pap tests) must enroll and participate in one testing event annually. If you only screen non-gynecologic cytology specimens, you do not need to enroll. *Participants are exempt from taking the PAP PT during the calendar year in which they pass their boards.
No, you have to pick only one of the available programs. If there is not a module that covers your specific slideset combination, please choose one that covers the majority of slides that your site interprets. PAPJPT may only be ordered by sites performing all 3 slideset types: Thin Prep, Sure Path, Conventional.
| Slide Type | Program Code | Individual Participant Response Form Code |
| Conventional | PAPCPT | APAPCPT |
| SurePath | PAPKPT | APAPKPT |
| ThinPrep | PAPMPT | APAPLPT |
| Combination: SurePath and ThinPrep | PAPLPT | APAPLPT |
| Combination: Conventional, SurePath and ThinPrep | PAPJPT | APAPJPT |
Call a Customer Contact Center Representative at 800-323-4040 or 847-832-7000. You will need to order a Locum Tenens examination. The Locum Tenens order can be used for pathologists or cytotechnologists who lack affiliation with a facility.
No. If you work at more than one laboratory, you will be required to select one laboratory as the primary site where you will be tested. All laboratory directors, however, must ensure that you participate in the required annual testing in order to meet their regulatory duties and these directors will also receive copies of your test results.
Yes. Any individual, including a Locum Tenens or temporary employee, who examines gynecologic cytology specimens must participate in annual testing and score at least 90%.
An extra test should be ordered if a participant missed the initial testing event with a valid reason (excused absence). The extra test is also the correct order for staff (pathologist or cytotechnologist) that joined the facility after its annual PAP PT was completed, and have not taken a PAP PT test at another site for the current year.
A retest is only ordered if a participant received less than 90% on their annual PAP PT examination and must take another test. Participants who transfer facilities and have taken a test previously during the year but have not passed must register for a retest.
Individuals who fail a test must remain with their originally selected PT program for that calendar year to complete any necessary re-testing.
It is best to have a proctor who is familiar with laboratory procedures. The CMS requires that each site setup at least 2 proctors. Examples of personnel that the Laboratory Director can designate as a proctor are the laboratory administrative assistant, a medical technologist, a histotechnologist, a quality supervisor or manager.
More detailed information can be found in the Cytopathology Topic Center at cap.org.
Refer to the Organizational Profile: How to Add/Delete Proctors/Examinees for the PAP PT Program.
DIAGNOSTIC IMMUNOLOGY AND FLOW CYTOMETRY
No. The FL6 program is designed for laboratories who are performing flow cytometry analysis on patients being treated with immunotherapy regimens (CAR-T and others).
No. The FL8 program asks participants to identify if MRD is present in mature B-Cell disorders. It is not limited to one disease type.
No. The BALL program allows for laboratories who are using COG and/or LDT assays to report.
In order to provide robust challenges to our customers, we use a cell line/whole blood mixture for the FL3 program. It may be beneficial to run unstained cells to assist in determining the antibody distribution of the population of interest.
For wet challenges, your laboratory only needs to report the antigens that it would for a patient. If you do not test an antigen, leave that section blank on the result form.
HEMOSTASIS
In a continuing effort to monitor the accuracy and reporting of the International Normalized Ratio (INR), the CAP has implemented an additional grading scheme for the INR. In addition to the grading scheme that is based upon the comparison of your laboratory’s INR to that of your peer group, a second evaluation will be provided. This evaluation compares your laboratory’s reported INR to a calculated INR that is based upon the International Sensitivity Index (ISI), the mean normal PT used to calculate the INR, and the PT result reported for each challenge that you provided on the result form. If you report the INR, but did not report the ISI, the mean normal PT and the PT on the result form, the summary will indicate unacceptable performance. Prothrombin time is a regulated analyte required under CLIA. The values that your laboratory reported might not have been sufficiently abnormal to cause a failing INR score in comparison with your peer group. However, a systematic error of the type detected and reported in this summary could be clinically significant, now and/or at the time of a change of reagents. As you know, it is extremely important for patient care to produce accurate INRs. Accordingly, the CAP strongly recommends that you examine the method used in your laboratory if your laboratory fails the special INR evaluation. For the special INR evaluation, there are several criteria required to determine the accuracy of the reported INRs. We must receive the PT INR results and reported INRs for at least four challenges in addition to the reported ISI and mean reference range PT. If fewer than four challenges are provided, we cannot assess the accuracy of the INRs and the result will be Insufficient Data for Evaluation. If at least 80% of the reported INRs are within ± 0.2 of the calculated INR, the result is Acceptable Performance.
HEMATOLOGY AND CLINICAL MICROSCOPY
Participants should reference the online Hematology Glossary before the Hematology Atlas as the glossary is updated yearly, whereas, the atlas is updated every 10 years. Participants should read the clinical histories which contain information to help them with their response(s). Identify the arrowed cell and not surrounding cells. No two image challenges will have the same master list identification.
Participants should read the clinical histories which contain information to help them with their platelet estimate response(s).
If you are enrolled in an automated hematology program that includes a manual differential (10 pictures), the manual differential takes precedence over the automated differential.
On test evaluations, reviewing the “Plot of the Relative Distance of Your Results from Target as Percentage of Allowed Deviation” that appears along the right-hand column can help determine if your instrument is experiencing either a positive or negative bias. Should this trend continue over 2 mailings, your instrument may need to be recalibrated.
HISTOTECHNOLOGY
A committee of pathologists and histotechnologists from the CAP and NSH determine which stains and tissues will be assessed in each challenge. Specific tissues and stains are selected to identify common problems (eg, fixation and processing of fatty breast tissue; optimal cytologic detail in decalcified bone marrow biopsies; proper embedding and sectioning of skin biopsies; or optimum preparation of amyloid stains). The committee strives to include cases that will allow laboratories to identify areas that can be improved.
The CAP/NSH Histotechnology Committee is pleased to offer a resource for laboratory staff to use for educational purposes and troubleshooting fixation, microtomy, and H&E staining technical problems. Participants of the HistoQIP programs that feature H&E staining can click Access e-LAB Solutions Suite, select their laboratory, then click Evaluation Reports to view the specific comments associated with their individual program.
By clicking on any comment link within your laboratory’s evaluation report, the online Digital Reference Tool (DRT) for H&E Staining will open to a web page that describes that problem, its cause(s) potential solutions, and digital images illustrating the artifact. Clicking on the thumbnail image will open DigitalScope, a digital microscope viewer, to show a stained microscopic image of the artifact, staining problem or technical issue. Each image is navigable in the same manner as a glass slide under a microscope, can be easily focused to a higher or lower magnification, and has annotation for that comment. There are multiple digital image examples associated with each artifact.
Note: The DRT does not address problems, causes, or potential solutions for IHC staining at this time.
The DRT can be used:
- As a learning exercise for laboratory staff to visualize exact problems reported in the HistoQIP evaluation report on the laboratory’s daily work, and then to use the troubleshooting guides to improve performance.
- As a competency assessment tool for laboratory staff who must complete annual demonstration of their technical knowledge and skills.
- As an educational enhancement for staff experiencing performance problems.
- As a method for delivering continuing education in a concise, meaningful format that directly correlates with laboratory bench duties.
- As a teaching tool for students and employees seeking to learn skills that can help them prepare for histotechnology certification.
IMMUNOHISTOCHEMISTRY
You may send out your PT/EQA slides for staining and digital analysis to a reference laboratory if that is in your laboratory procedures and as long as the reference laboratory and their software is not quantitating or interpreting the results.
INSTRUMENTATION
If your evaluation is nonlinear, but your results are similar to your peer group, the nonlinearity may be inherent in the method and is not specific to your instrument. The nonlinearity may also be due to a matrix effect in the calibration verification/linearity (CVL) material. It may be helpful to review the Peer Results Summary Table included in your evaluation report to further assess peer group performance.
The linearity evaluation is based solely on your laboratory’s results. The calibration verification compares your results to a peer group mean. Several factors such reporting wrong unit of measure, incorrect specimen handling, and differing lots of reagents and calibrators can contribute to a different evaluation.
MICROBIOLOGY
The United States federal government regulations require that culture challenges be a mixture of report all challenges and principle pathogen challenges. If the PT/EQA kit instructions or result form say to report all organisms, laboratories are to report everything that grows, even organisms their laboratory would not typically report because it might be considered normal flora. If the PT/EQA instructions indicate to report principle pathogen(s), laboratories should report the organism(s) that are causing the infection. There are two sets of reporting boxes available on the result form and a laboratory can report up to two organisms. If the PT/EQA challenge contains an organism that would be pathogenic for the source and another organism that would be normal flora, participants are expected to only report the pathogen. Do not report the normal flora organism.
Laboratories should report PT/EQA to the level they report patient results. Each organism is graded individually, and all correct responses are considered good.
Laboratories should report only the testing they perform on the specimen. For example, if the glutamate dehydrogenase (GDH) antigen is negative and you would not test for the toxin, you should report the toxin as, “test not performed.” GDH is regulated as bacterial antigen and can be sent to CMS. If it is on a laboratory’s Analyte Reporting Selection (ARS), then GDH “test not performed” must be selected for all specimens if no testing for that analyte is performed.
No interpretation (NI) should only be used if Standards Development Organizations (SDO) do not have breakpoint interpretations. If your institution reports only the minimum inhibitory concentration (MIC) but an interpretation is available from the SDO, NI will be considered a penalty. Since only interpretations are graded, do not report only the antifungal agent and MIC value. If the antifungal agent is entered and an MIC is entered, an interpretation must be entered or a penalty will occur.
The D program material is live organisms lyophilized on a swab therefore it should work for molecular or enzyme immunoassay (EIA) testing; however, the D program is intended for culture use. There are no specific testing instructions for these procedures. CMS does allow bacterial identification by molecular methods.
Each GDH antigen and toxin test includes a method reporting area. Laboratories must ensure they enter the correct method. If multiple methods are tested for one analyte per laboratory protocols (ie, toxin initially tested by nucleic acid amplification [NAA] method and followed up by antigen) then the last and final method/result should be reported for PT/EQA.
A laboratory cannot use more than one method for a specimen. This would be considered as duplicate testing. For PT/EQA, laboratories should use the primary method used for patient testing. If more than one method may be used, a laboratory can rotate mailings between methods.
The D1 (Throat Culture) program can be tested by molecular methods.
Specific manufacturer detailed instructions that are included in the kit instructions come directly from that manufacturer. If no instructions are included for your manufacturer, it is assumed to test as a patient specimen, or contact the manufacturer to see if they can give you any specific instructions.
MOLECULAR ONCOLOGY
Enrollment in NGSST would be required for CAP-accredited laboratories.
Enrollment in NGSHM would be required for CAP-accredited laboratories. It is not necessary to also enroll in MHO program for this assay.
QUALITY PRACTICES
For certain metrics to be shown in your quality management program reports, you must complete the Quality Management Tools Demographics (QDEM) form on e-LABS Solutions Suite.
To access the online QDEM result form:
- Scroll to the top of this page and click Access e-Lab Solutions Suite.
- Click View, enter, or submit PT results. Enter your CAP user ID and password if you are prompted to do so.
- Open the Filter Option(s) section, enter QDEM-A into the Mailing(s) starting with field, and click Go.
- To begin reporting results, click Enter Data in the Data column.
Additionally, for QT programs, you must also complete and return the Customer-Defined Comparison Group input form with the first data submission of the year, even if your selections are the same as last year. Enter codes that you would like to use for comparison using the Customer-Defined Comparison Group Master List in the kit instructions. Once you enter the codes for the year, you do not need to re-enter them during the calendar year unless you need to make changes.
You can correct your data or add new data to your QP result form up to 8 weeks after the original publication of the Participant/Preliminary Report or Participant/Preliminary Report and Data Analysis critique (report style dependent on the program). To do this, call the contact center to extend your due date. New reports will be issued online approximately 9 weeks after the original publication date.
You can correct your data or add new data to your QT result form(s) going back three quarters (eg, during the D mailing period, you can go back to the A, B, or C mailing to add or correct your data. The new or edited data will show in your next quarterly report. To add data after the current mailing due date or to go back to a prior mailing, call the contact center to extend your due date. It is best to look at your reports for missing data and errors within 6 weeks of receipt; if the CAP has made a processing error, a new report will be issued.
Only report up to 120 critical values for inpatient and 120 critical values for outpatients, up to 240 total.

REPRODUCTIVE MEDICINE
Sperm viability assessment on PT/EQA specimens should be performed in the same manner in which patient specimens are assessed. For additional information, consult the most up to date semen analysis manual for additional information such as the WHO laboratory manual for the Examination and processing of human semen, 6th edition.
Instructions are provided online via the “View Video(s)/Image(s)” link, which provide calculations and examples based on the type of chamber video you are viewing.
SURGICAL PATHOLOGY
The catalog is created so far in advance that it is challenging to define a focus.