- Home
- Member Resources
- Articles
- CAR T-Cells Therapies: Opportunities and Challenges

Figure 1: Schematic structure of a chimeric antigen receptor (CAR) (Cancer Sci 108 [2017] 1109–1118. CC-BY-NC-4.0)
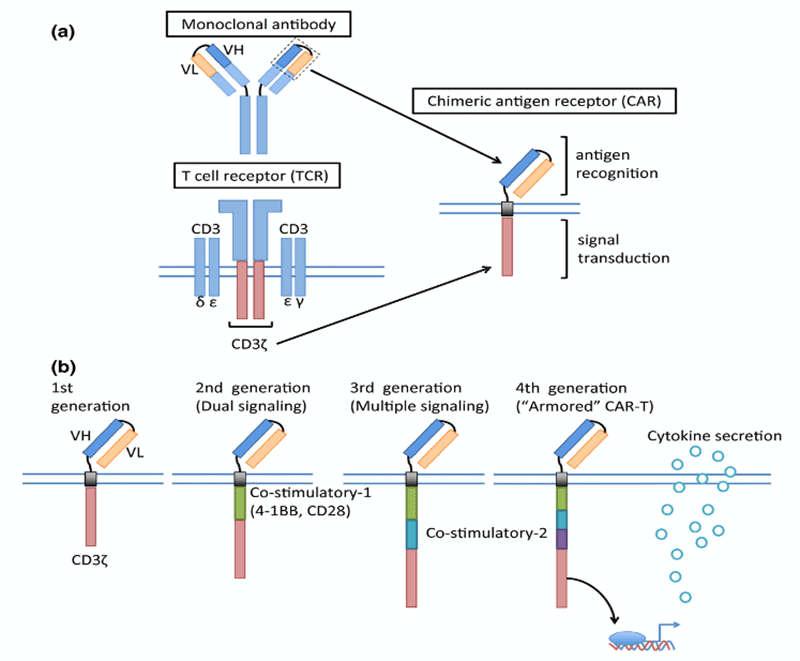
Chimeric antigen receptor (CAR) T-cell therapy is one of the most promising approaches in anticancer therapy and an exponential number of pre-clinical and clinical studies are being carried out every year to expand its use. CAR T-cell therapy represents a cellular immunotherapy where T-cells are redirected against the tumor after engineered expression of CARs. CARs are receptor proteins that have been designed to give T-cells the new ability to target a specific antigen. The process involves taking T-cells from blood, editing the T-cells to express a CAR to recognize a specific antigen on the targeted cell (e.g., CD19 on B-cells or CD38 on plasma cells), growing those cells in the laboratory, and then administering them to the patient to bind to cancer cells and eliminate them.
CAR constructs consist of four main components: (1) an extracellular target antigen binding domain, derived from the variable heavy (VH) and variable light (VL) chains of monoclonal antibodies, connected via a flexible linker to form a single chain variable fragment (scFv); (2) a hinge or spacer region that provides flexibility; (3) a transmembrane domain that anchor the CAR to the T cell membrane, and (4) one or more intracellular signaling domains (FIGURE 1). First generation CARs engineered in the late 1990s contained a CD3ζ or FcRγ signaling domain.1,2 With the improved understanding of the importance of co-stimulation for durable CAR-T cell therapy, second generation CARs with one co-stimulatory domain in series with the CD3ζ intracellular signaling domain were generated. The two most common co-stimulatory domains CD28 and 4-1BB (CD137) are both associated with high patient response rates.2-4 Since co-stimulation through only one domain produces incomplete activation, third generation CARs, which incorporate two costimulatory domains in series with CD3ζ have been investigated in preclinical studies, showing mixed results.5 The CAR constructs are transduced into T-cells isolated from the patient using leukapheresis via a gammaretrovirus or a lentiviral vector, and subsequently re-infused into the patient.
Tisagenlecleucel (Kymriah, Novartis Pharmaceuticals), an autologous anti-CD19 CAR T-cell therapy, was the first CAR T-cell therapies approved by United States Food and Drug Administration (FDA) in 2017 for management of patients with relapsed or refractory B-cell precursor acute lymphoblastic leukemia (BALL) and later in 2018 for diffuse large B-cell lymphoma (DLBCL), high-grade B-cell lymphoma (HGBCL), and DLBCL arising from transformed follicular lymphoma (tFL). The results of the JULIET trial in DLBCL6 showed the best objective response rate (ORR) was 52%, with 40% of patients achieving complete response (CR). At 32.6 months of follow-up, the median duration of response (DOR) was not reached, and median overall survival (OS) was 11.1 months. Axicabtagene ciloleucel (Yescarta, Kite Pharma) was also approved later in 2017 for large B cell lymphoma including DLBCL, primary mediastinal B-cell lymphoma (PMBCL), HGBCL, and tFL. In the pivotal ZUMA-1 phase 1/2 clinical trials,7 the best ORR was 74% with 54% CR; the reported median time to best response was 1 month. The median DOR was 11.1 months, median OS was not reached, and the median progression-free survival (PFS) was 5.9 months.
Lisocabtagene maraleucel (Breyanzi, Juno Therapeutics), another autologous CD19-directed CAR T-cell therapy, was FDA approved in 2021 for patients with large B-cell lymphoma (LBCL), including DLBCL, HGBCL, PMBCL, and follicular lymphoma grade 3B. The TRANSCEND trial showed best ORR of 73%, with a CR rate of 53%, while the median OS and PFS were 21.1 months and 6.8 months, respectively.8 In addition to the ZUMA-1, JULIET, and TRANSCEND trials, several other studies have shown similar outcomes in terms of ORR and OS of CD19-targeted CAR T-cell therapies.9,10 Currently, six commercially produced CAR T-cell therapies have been approved by the FDA for management of aggressive hematologic malignancies (Table 1). Moreover, more than 400 clinical trials are ongoing worldwide to evaluate the efficacy of different CAR T-cell constructs in a range of hematologic malignancies, as well as their use in pediatric and adult solid tumors.11
Despite their success, there are major limitations to CAR T-cell therapy that still must be addressed, including life-threatening CAR T-cell associated toxicities (cytokine release syndrome, neurologic toxicities, prolonged cytopenias, etc.), inhibition and resistance in B-cell malignancies, antigen escape, limited persistence, poor trafficking and tumor infiltration, immunosuppressive microenvironment, limited efficacy against solid tumors, and the spector of secondary malignancies.2,12 However, one of the most challenging limitations is accessibility, since only 20% of the patients for which CAR T-cells therapy is indicated have access to the treatment.13 The disadvantages affecting accessibility are linked to the autologous nature of the therapy, and include the cost of the process, the manufacturing failure in some patients, and the current prolonged manufacturing processes of approximately three weeks, which translate into a delay in the availability of the treatment. For these reasons, there is growing interest in the development of allogeneic cellular therapies where the T-cells are obtained from healthy donor cells rather than from patients, a so-called “off-the-shelf” option. 14,15
Allogeneic products present several advantages. First, a higher number of CAR T-cells can be produced from a single donor, with simplification and standardization of the products based on donor selection and processing. Moreover, allogeneic CAR T-cells may also allow for combinations directed against multiple targets. Cryopreservation of batches of T-cells allows for immediate availability for treatment, and also for redosing if needed. Finally, a decrease in costs is expected due to implementation of industrialized manufacturing processes.16 However, allogeneic approaches are associated with major issues, such as cytokine release syndrome; CAR and/or gene editing-related gene modifications; rejection of allogeneic cells; graft-versus-host disease (GVHD); toxicity in the case of intense lymphodepletion. Among these issues, the latter two, GVHD and T cell depletion, represent the main obstacles, primarily because administered allogeneic T cells may cause life-threatening GVHD, and secondarily because the allogeneic T cells may be rapidly eliminated by the host immune system, limiting their anti-tumoral activity. Thanks to the development of gene editing technologies, the risk of GVHD has been reduced by efficiently eliminating TCR expression, and new techniques have been generated to make allogeneic CAR T cells “less visible” to the host immune system.15
The initial success of CAR T-cell therapy has led to the expansion of study of other forms of adoptive cell therapies, with natural killer (NK) cells being one of the most promising other options. NK cells recognize “non-self” cells without the need for antibodies and major histocompatibility complex (MHC), causing a rapid immune reaction and rapid killing. The generation of CAR constructs for CAR NK-cells is similar to CAR T-cells, but the costimulatory domains have greater specificity for NK cell signaling, such as DAP10, DAP12 and 2B4.17 CAR NK-cells therapies were first tested for hematologic malignancies, and more data on their use against solid tumor are being generated, with several clinical trials currently ongoing, including CAR NK-cells targeting solid tumors such as epithelial ovarian cancer, prostate cancer, and glioblastoma.11 The favorable safety profile and the relatively large proportion of NK cells in the lymphocyte population make NK cells one of the best candidates for allogeneic adoptive immunotherapy. Many sources of NK cells have been used to produce them, such as NK-92 cell line, NK cells derived from umbilical cord or peripheral blood, and NK cells derived from induced pluripotent stem cells (iPSCs). Indeed, the first “ready to use,” universal and allogeneic CAR NK cellular product derived from iPSCs (FT596) was cleared in 2022 by the FDA for the treatment of B cell lymphoma and chronic lymphocytic leukemia.18
Advances in CAR T-cells therapies (as well as other cellular therapies) has fueled growth and expansion of transfusion medicine. Leukapheresis (white blood cell separation and collection using apheresis) is a pivotal procedural step in CAR T-cell production. Apheresis practitioners provide oversight for leukapheresis and ensure optimal product is collected for CAR T-cell manufacturing (purification, CAR engineering, and expansion). Unfortunately, there is lack of standardization of protocols for apheresis collections.19 Sponsors of CAR T-cell therapies (pharmaceutical companies and clinical trials) usually have their own protocols for how cells should be collected, labeled, and transported. Increased demand and lack of standard collection protocols places undue pressure on apheresis units and can compromise quality of product collected. It is high time to standardize apheresis collections to ensure patient access, expanding coverage for transformative therapies, advancing novel biotherapies research, and increasing the supply and safety of biological products. Methodologies to monitor the quality and quantity of CAR T-cells before and after infusion are also urgently needed.
Following almost three decades of development and initial success, further expansion of CAR T-cell therapies is inevitable. Future of cellular therapies will not only include autologous and allogeneic products but exploring efficacy in indications other than hematologic malignancies and novel in-vivo CAR T-cell gene therapies. After initial failure, few success stories of CAR T-cell therapy in solid tumors like pancreatic cancer and non-small cell lung cancer have been reported and clinical trials are underway to support feasibility and safety in treating solid tumors.20-22 Various pathology and laboratory medicine services including hematopathology, flow cytometry, molecular pathology, and transfusion medicine to name a few are involved in providing diagnostic and therapeutic care to the patients requiring CAR T-cell therapy and need to be trained to recognize the effects and side effects of this treatment. The pathology workforce must adapt to meet the needs of this growing and evolving field.2 We should continue to nurture close collaboration with oncologists and develop educational programs to train our workforce to make these treatments a reality for patients.
| PRODUCT | INDICATIONS | TARGET (CO-STIMULATORY MOLECULE) | TYPE |
|---|---|---|---|
ABECMA® (idecabtagene vicleucel) |
Relapsed/refractory multiple myeloma |
BCMA (4-1BB) |
CAR-T |
BREYANZI® (lisocabtagene maraleucel) |
Adults: Relapsed/refractory large B-cell lymphoma |
CD19 (4-1BB) |
CAR-T |
CARVYKTITM (ciltacabtagene autoleucel) |
Relapsed/refractory multiple myeloma |
BCMA (4-1BB) |
CAR-T |
KYMRIAHTM (tisagenlecleucel) |
Adults: Relapsed/refractory diffuse large B-cell lymphoma (DLBCL) Children: Relapsed/refractory acute lymphoblastic leukemia (ALL) |
CD19 (4-1BB) |
CAR-T |
TECARTUSTM (brexucabtagene autoleucel) |
Relapsed/refractory mantle cell lymphoma. Relapsed/refractory B-cell precursor acute lymphoblastic leukemia (ALL) |
CD19 (CD28) |
CAR-T |
YESCARTATM (axicabtagene ciloleucel) |
Refractory/relapsed: Diffuse large B-cell lymphoma (DLBCL) Primary mediastinal B-cell lymphoma High grade B-cell lymphoma DLBCL that results from follicular lymphoma Follicular lymphoma |
CD19 (CD28) |
CAR-T |
Table 1. Current FDA approved CAR products
References
- Gross G, Waks T, Eshhar Z. Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc Natl Acad Sci U S A. 1989;86(24):10024-28.
- Cushman-Vokoun AM, Voelkerding KV, Fung MK, et al. A primer on chimeric antigen receptor T-cell therapy: what does it mean for pathologists?. Arch Patho Lab Med. 2021; 145(6): 704-16.
- Maher J, Brentjens RJ, Gunset G, Rivière I, Sadelain M. Human T-lymphocyte cytotoxicity and proliferation directed by a single chimeric TCRζ/CD28 receptor. Nat Biotechnol. 2002;20(1):70-75.
- Imai CM, Mihara K, Andreansky M, Nicholson IC, Pui CH, Geiger TL, Campana D. Chimeric receptors with 4-1BB signaling capacity provoke potent cytotoxicity against acute lymphoblastic leukemia. Leukemia. 2004;18(4):676-84.
- Zhong XS, Matsushita M, Plotkin J, Riviere I, Sadelain M. Chimeric antigen receptors combining 4-1BB and CD28 signaling domains augment PI3kinase/AKT/Bcl-XL activation and CD8+ T cell–mediated tumor eradication. Mol Ther. 2010;18(2):413-20.
- Schuster SJ, Bishop MR, Tam CS, Waller EK, Borchmann P, McGuirk JP, et al. Tisagenlecleucel in adult relapsed or refractory diffuse large B-cell lymphoma. N Engl J Med. 2019;380(1):45-56.
- Locke FL, Ghobadi A, Jacobson CA, Miklos DB, Lekakis LJ, Oluwole OO, et al. Long-term safety and activity of axicabtagene ciloleucel in refractory large B-cell lymphoma (ZUMA-1): a single-arm, multicentre, phase 1–2 trial. Lancet Oncol. 2019;20(1):31-42.
- Abramson JS, Palomba ML, Gordon LI, et al. Lisocabtagene maraleucel for patients with relapsed or refractory large B-cell lymphomas (TRANSCEND NHL 001): a multicentre seamless design study. Lancet Oncol.2020;396(10254):839-852.
- Jacobson CA, Hunter B, Armand P, Kamihara Y, Ritz J, Rodig SJ, et al. Axicabtagene ciloleucel in the real world: outcomes and predictors of response, resistance and toxicity. Blood. 2018;132:92.
- Pasquini MC, Locke FL, Herrera AF, Siddiqi T, Ghobadi A, Komanduri KV, et al. Post-marketing use outcomes of an anti-CD19 chimeric antigen receptor (CAR) T cell therapy, axicabtagene ciloleucel (Axi-Cel), for the treatment of large B cell lymphoma (LBCL) in the United States (US). Blood. 2019;134:764.
- US National Institutes of Health. ClinicalTrials.gov. www.clinicaltrials.gov/.
- Sterner RC, Sterner RM. CAR-T cell therapy: current limitations and potential strategies. Blood Cancer J. 2021;11(4):1-11.
- Borgert R. Improving outcomes and mitigating costs associated with CAR T-cell therapy. Am J Manag Care. 2021;27(13 Suppl):S253-61.
- Zhao J, Lin Q, Song Y, Liu D. Universal CARs, universal T cells, and universal CAR T cells. J Hematol Oncol. 2018;11(1):1-9.
- Depil S, Duchateau P, Grupp SA, Mufti G, Poirot L. ‘Off-the-shelf’ allogeneic CAR T cells: development and challenges. Nat Rev Drug Discov. 2020;19(3):185-99.
- Morgan MA, Büning H, Sauer M, Schambach A. Use of cell and genome modification technologies to generate improved “off-the-shelf” CAR T and CAR NK cells. Front Immunol. 2020;11:1965.
- Schmidt, P.; Raftery, M.J.; Pecher, G. Engineering NK Cells for CAR Therapy—Recent Advances in Gene Transfer Methodology. Front Immunol. 2021, 11, 61163.
- Sabbah, M. Jondreville, L. Lacan, C, etal. CAR-NK Cells: A chimeric hope or a promising therapy? Cancers (Basel). 2022, 14(15):3839.
- Horne GA, Laird J, Latif AL, Irvine D, Wilson M, Douglas K. The implications of CAR-T cell therapy on apheresis services: a Scottish perspective. J Clin Apher. 2021;36(3):513-5.
- Newick K, O'Brien S, Moon E, Albelda SM. CAR T cell therapy for solid tumors. Annu Rev Med. 2017;68:139-52.
- Marofi F, Motavalli R, Safonov VA, et al. CAR T cells in solid tumors: challenges and opportunities. Stem Cell Res Ther. 2021;12(1):1-6.
- Ma S, Li X, Wang X, Cheng L, Li Z, Zhang C, Ye Z, Qian Q. Current progress in CAR-T cell therapy for solid tumors. Int J Biol Sci. 2019;15(12):2548.

Valentina Zanfagnin, MD, is a third-year Pathology resident at the University of Southern California, Los Angeles. Dr. Zanfagnin earned her Medical Degree from the University of Turin, Italy, and completed a post-doctoral fellowship in Translational Medicine at the Mayo Clinic of Rochester, MN. Her research interests focus on the molecular characterization of solid tumors, biomarkers discovery and tumor microenvironment characterization. She will pursue a Surgical Pathology Fellowship at the Massachusetts General Hospital in Boston, with a focus on perinatal, gynecologic and breast pathology.

Gagan Mathur, MD, MBA, CPE, FCAP, is a board-certified transfusion medicine/blood banking physician and serves as the medical director of transfusion medicine at the University of California, Irvine (UCI). Dr. Mathur holds the academic title of associate professor of clinical pathology in the Department of Pathology & Laboratory Medicine at the UCI School of Medicine. He pursued his master of business administration (MBA) from the University of Iowa Tippie School of Management, along with his clinical training in clinical pathology at the University of Iowa Hospitals & Clinics. Dr. Mathur earned the title of Certified Physician Executive from American Association for Physician Leadership. Dr. Mathur’s areas of clinical interest and expertise include patient blood management, therapeutic apheresis, and cellular therapies. Along with his clinical responsibilities, Dr. Mathur is enthusiastically involved in clinical research and teaching to accomplish his academic commissions. Utilizing his interest and training in business management, informatics, and healthcare administration, Dr. Mathur strives to provide effective and efficient patient care.