- Home
- Member Resources
- Clinical Informatics Resources
- Don’t Forget Your Rules When Harmonizing Laboratory Testing Across Multiple Sites
Read the cases in order starting with Part 1. The case includes questions to support your learning as you progress through each part. In addition, the case includes a summary, key points, multiple-choice questions with answers, and a list of references. After completing at least one case (hopefully you will complete all three), please provide your feedback via a short survey.
- Open all
- Close all
Dr. Feingold, the head of the infection control committee in charge of harmonizing practices at three of the local hospitals in your health care system tells you, the new medical director responsible for laboratory harmonization, that he would like to set up a standardized protocol for reflexively evaluating high risk urinalysis samples by urine culture because urine cultures were being over-utilized and misutilized. In fact, a recent survey of nursing staff and resident physicians in the emergency departments and intensive care units at each of the sites showed that urine cultures were sometimes being ordered for inappropriate and non-evidence-based reasons such as “urine having a strong smell” or urine in a Foley “looking like it’s infected.”
He asks if you can help the team to create a single reflex order in the laboratory information system (LIS) so that urinalysis specimens will be treated uniformly across all sites. If the macroscopic and microscopic results of the urinalysis meet predefined diagnostic criteria, then those specimens will automatically be reflexed to urine culture. You agree that individual orders for urine culture or urinalysis will still be available for specialized services, so that clinicians who require those specific tests results will still be able to order the tests individually.
You tell Dr. Feingold that the department of laboratory medicine will be happy to help set up the reflex testing since it will improve patient care and test utilization by reducing the number of cultures which are currently being performed for inappropriate reasons. You agree to set up a urinalysis with reflex to urine culture testing order for deployment across all three hospital laboratories. After meeting with the supervisors of the microbiology and chemistry laboratories at the three sites, you learn that the same urinalysis instruments are being used in all three laboratories. You request the standard operating procedures (SOPs) for urinalysis from all three sites and a sample of how the results appear when they are reported in the electronic health record (EHR).
SOP and Sample of EHR Results
 | 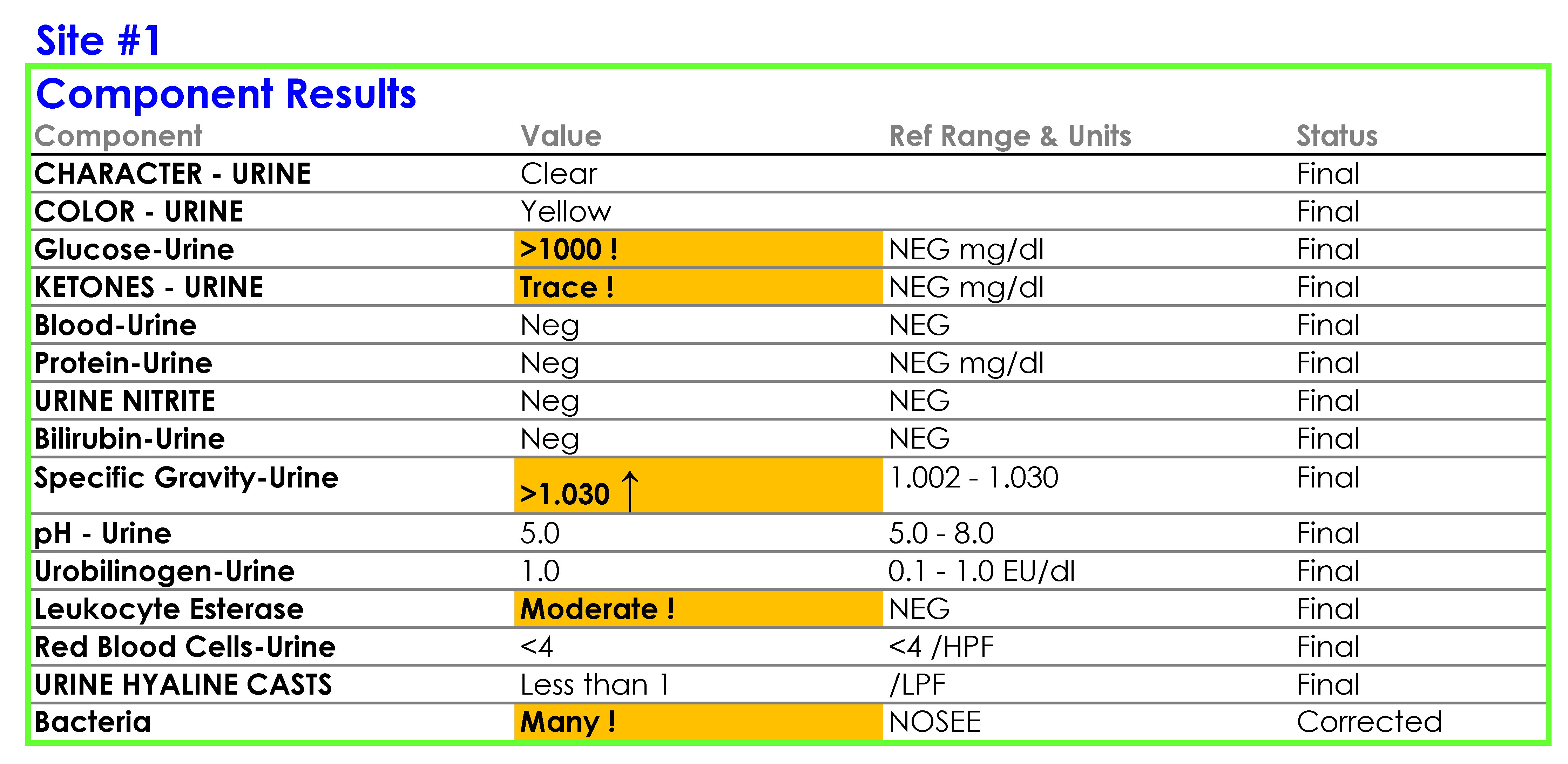 |
 | 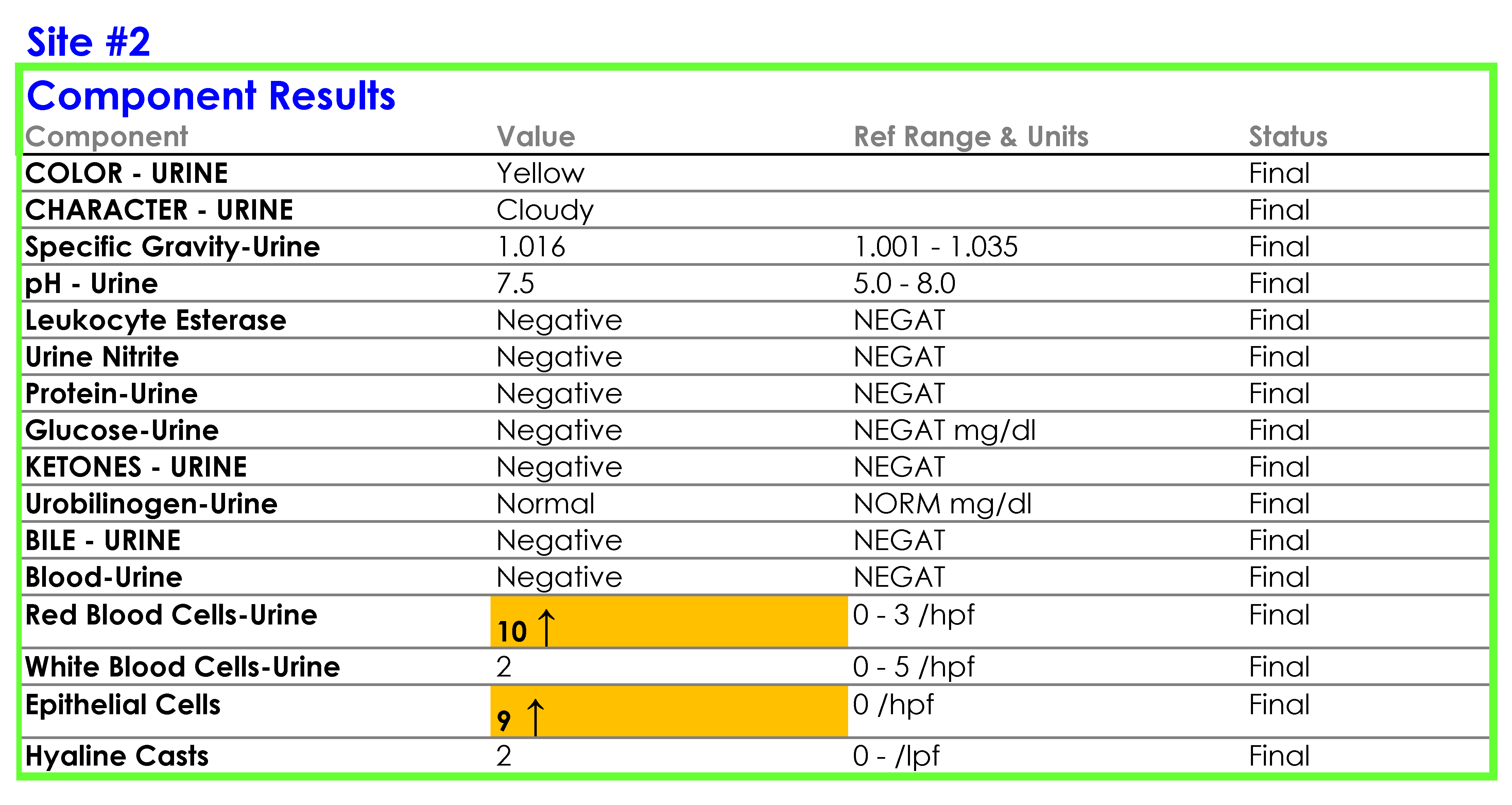 |
 | 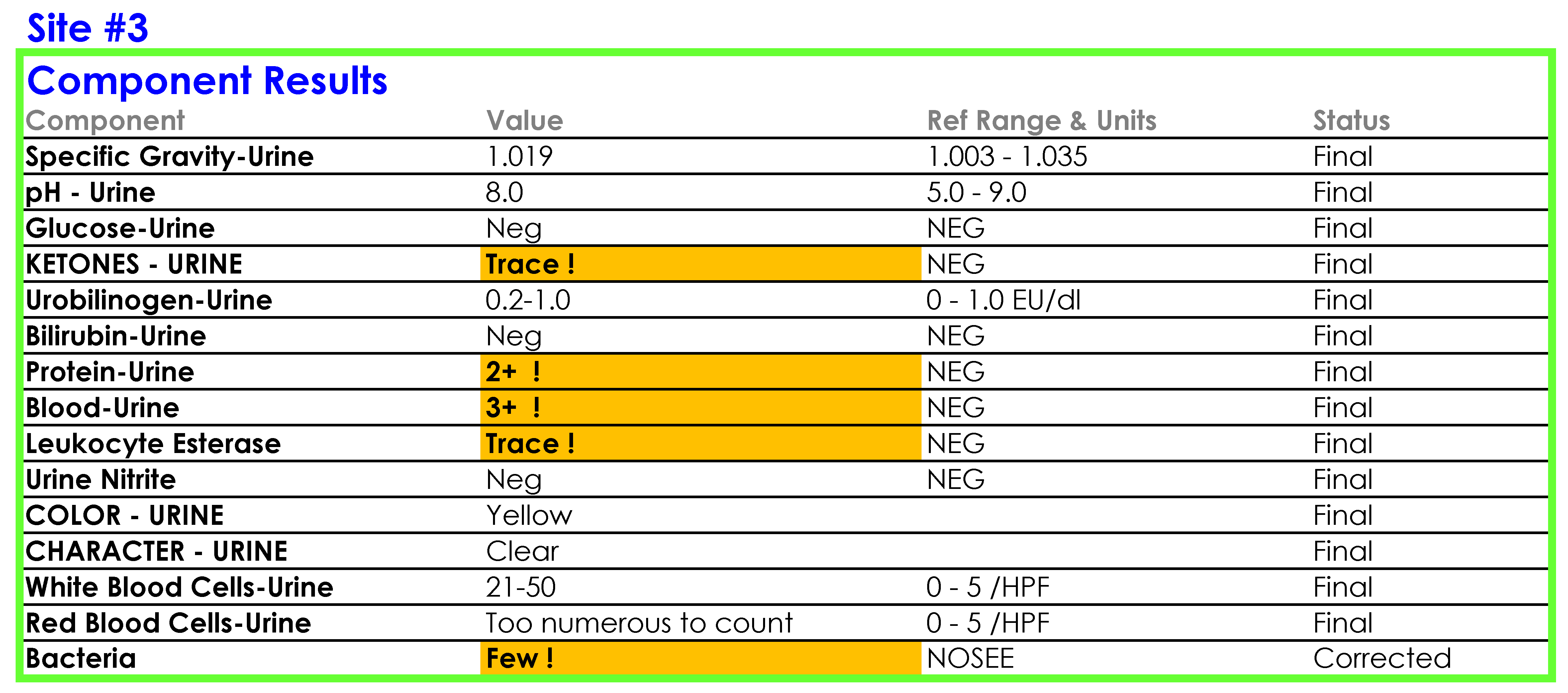 |
Questions to Think About
1. What role does reflex testing play in the laboratory?
Reflex testing is an important tool for directing additional testing without the intervention of the ordering physician. Reflex testing occurs based on a predefined set of rules, such as when an initial test result meets specified criteria (e.g., positive or outside reference parameters). The purpose of reflex testing is to provide timely, cost-effective follow-up testing to guide care which cannot be determined solely by the results of the primary test.
2. Why would it be important to have the director and laboratory staff review the SOPs of all sites involved in harmonization?
Harmonizing laboratory practices across sites requires a review of each laboratory's current practices and a discussion about appropriate or relevant differences (such as different patient populations served). The laboratory director and managers need to have a high level view of the differences and similarities of individual laboratory practices with an aim to optimize the performance of testing, align laboratory practices when possible, and ensure that everyone is cognizant of the variations in practice, especially when considering the implementation of reflex testing with uniform rules across multiple sites.
You notice a number of discrepancies between the SOPs of the three laboratories, potential errors in the SOPs themselves, and problems with how the results are being reported in the LIS and EHR, some of which are a consequence of the middleware rules set up by each laboratory, for example:
For Site #1, the SOP stated that white blood cells (WBC) were enumerated as WBCs per high power field (HPF); however, you noticed that a WBC within the reference range (0-5 /HPF) was being hidden from reporting and did not appear in the EHR results.
For Site #2, the SOP stated that bacteria were reported as “None” if they were not present; however, you found that a negative urine bacteria was being hidden from reporting and did not appear in the EHR results.
For Site #3, the SOP stated that the reference range for red blood cells (RBCs) was 0-3 /HPF which did not correspond to the reference range in the EHR.
You ask the laboratory supervisors if their sites used middleware to connect the analyzers with the laboratory information system (LIS) and if they had rules setup for tests such as automated reflex testing and autoverification. They confirmed that each of the three laboratories were using reflex rules housed within each laboratory’s installation of the same middleware software purchased directly from the vendor.
You also confirm with the vendor that the raw data (original values on the analyzer), such as bacteria count in number/µL, were not being transmitted to the LIS. Therefore, the only result contained in the LIS (and by extension, the EHR) was the interpretation of the result by the middleware rules.
On a more thorough examination, you find that the middleware rules are not subjected to document control or review as part of the laboratories’ annual SOP review process and that the laboratories did not have a defined process for evaluating whether the results appeared in the EHR as expected. You ask the laboratory supervisors at each of the sites to provide you with the middleware rules. The pathology resident on service is surprised to learn that the three laboratories are all using the same analyzer and middleware and volunteers to make a table summarizing how each site is interpreting and reporting urinalysis results.
Questions to Think About - Part A
1. What is middleware?
Middleware is software that serves as an automation tool that connects different applications/hardware devices that need to interact with each other, such as connecting an analyzer with the laboratory information system.
2. What is the function of rules?
Rules are a defined, standard mechanism used to trigger a set of actions based on simple or complex formulas. Rules typically use one of the following syntax:
- IF condition(s) THEN action(s)
- IF condition(s) THEN action(s) ELSE action(s)
3. Why might it be valuable to include the middleware rules in the SOP?
Including the rules in the SOP makes the rules visible to everyone in the laboratory and helps to ensure that the values and interpretations in the SOP correspond with the rules being used to result patient samples.
4. Why would it be important to ensure that the middleware rules are reviewed as part of the annual SOP review process?
The rules are often not immediately accessible by the laboratory staff and are not necessarily something that staff have familiarity with despite the critical role the rules play in the laboratory’s workflow. By including the rules in an annual SOP review process, the laboratory staff have an opportunity to review the practices delineated in the rules in order to ensure that if changes are made to the SOP, those changes are reflected in the middleware rules.
5. One of the laboratory supervisors tells you that this discussion about middleware rules reminds her of the rules they have setup for autoverification. She would like to know if there is a CAP accreditation requirement for autoverification rules which the laboratory could use as guide for validating reflex testing rules. Which CAP checklist item would you review?
GEN.43875 Autoverification Validation Phase II
- There is documentation that the autoverification process was validated initially and is tested at least annually and whenever there is a change to the system that could affect the autoverification logic.
- NOTE: The range of results for which autoverification is acceptable must be defined for all patient tests subject to autoverification.
- Validation of autoverification must include a process to confirm that the autoverification algorithm decision rules are functioning properly, including the use of previously assayed specimens with results that challenge the algorithm. Examples of specimens that may be needed to validate the autoverification algorithm decision rules may include specimens with analyte concentrations within the normal reference limit, above or below the reference limits, above or below the analytic measurement range, and in the critical range. Specimens with known interferences and specimens that require calculations should also be used, when applicable.
- When changes are made that might affect the autoverification decision algorithm, validation appropriate to the scope and nature of the change must be performed.
- Evidence of Compliance:
- Records of autoverification validation studies approval AND
- Records of ongoing retesting of the autoverification process at least annually and at changes to the system.
- Source: College of American Pathologists. Laboratory General Checklist. Northfield, IL: College of American Pathologists; 2018.
6. According to the CAP, is the laboratory responsible for ensuring that the results appear in the EHR as expected? Which CAP checklist item mentions this responsibility?
GEN.48500 Interface Result Integrity Phase II
- There is a procedure to verify that patient results are accurately transmitted from the point of data entry (interfaced instruments and manual input) to patient reports (whether paper or electronic).
- NOTE: Verification must be performed prior to implementation of an interface (i.e. pre go-live) and whenever any change is made to an existing interface that could affect the accuracy of transmission of patient results. In addition, it must be reverified at least every two years. This includes evaluation of data transmitted from the LIS to other computer systems and their output devices.
- Verification of accurate data transmission from the LIS to other systems must be performed by reviewing data in the first downstream (or interfaced) system in which the ordering clinician/client (e.g. referring laboratory) may be expected to routinely access patient data.
- If the LIS has separate interfaces to multiple receiving systems in which patient data can be accessed by clinicians, then reports from each receiving system must be validated.
- However, where multiple sites use the same recipient system (e.g. the same installed instance of an electronic medical record system), validation need only occur for the interface (i.e. at one of the sites) and not for each individual site that is served by that single installed system.
- Interface validation should include:
- Examples of individual results, test packages or batteries, abnormal flags, and results with reference intervals and comments/footnotes.
- Initial interface validation should include:
- Verification that corrected results for clinical laboratory and anatomic pathology results are handled accurately in the receiving system.
- Evidence of Compliance:
- Printed screen shots or other verification records
- Source: College of American Pathologists. Laboratory General Checklist. Northfield, IL: College of American Pathologists; 2018.
Questions to Think About - Part B
Review the middleware rules below for the 3 sites and then answer the questions provided for each site.



Questions for Site #1
1. What result would you expect to see in the EHR for a WBC count of 1 to 5 per HPF?
The result for WBCs will be hidden and will not appear in the EHR.
2. What is the upper threshold for calling bacteria negative?
Bacteria less than or equal to 600 bacteria per µL.
3. What result would you expect to see in the EHR for a value of 3000 bacteria per µL?
The result for bacteria would be “Many.”
Questions for Site #2
4. What result would you expect to see in the EHR for a WBC count of 900 per HPF?
The result for WBCs will be reported as “Greater than 900.
5. What result would you expect to see in the EHR for a value of 500 bacteria per µL?
The result for bacteria will be hidden and will not appear in the EHR.
6. What result would you expect to see in the EHR for a value of 3000 bacteria per µL?
The result for bacteria would be “P2” (2+).
Questions for Site #3
7. What result would you expect for a value of 5 white cells per HPF?
The result for WBCs would be 5 /HPF.
8. What result would you expect to see in the EHR for a value of 3000 bacteria per µL?
The result for bacteria would be “Few.”
9. Based on the copy of the rules the supervisor provided you, can you tell how a bacteria count of 500, 3500, and 5000 /µL would be categorized/reported?
No. As written, the rules say that a bacteria count <=500 and a value of >=500 would be categorized as negative and rare, respectively; a bacteria count <=3500 and a value of >=3500 would be categorized as few and moderate, respectively; and a bacteria count <=5000 and a value of >=5000 would be categorized as moderate and many, respectively.
Review Tables 1, 1a, and 1b and then finish Part 3.
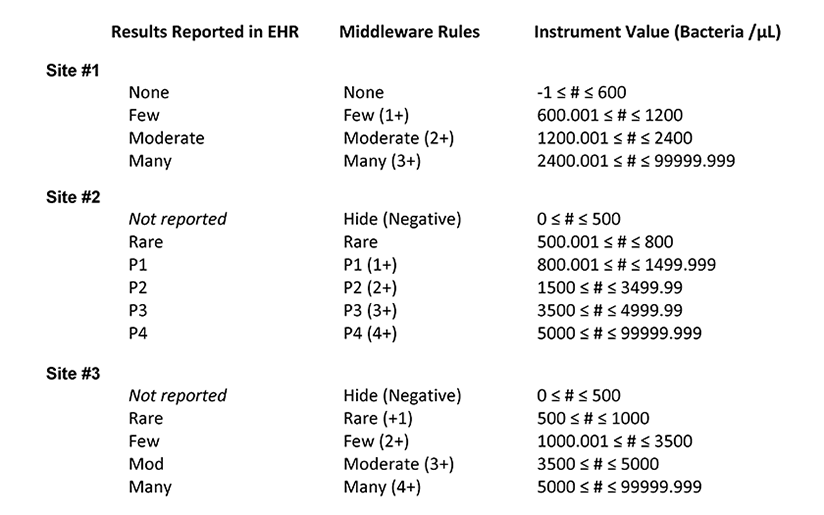 Table 1. Summary of the middleware interpretation of the instrument values for number of bacteria per µL and the corresponding results reported in the electronic health record (EHR) for each site
Table 1. Summary of the middleware interpretation of the instrument values for number of bacteria per µL and the corresponding results reported in the electronic health record (EHR) for each site
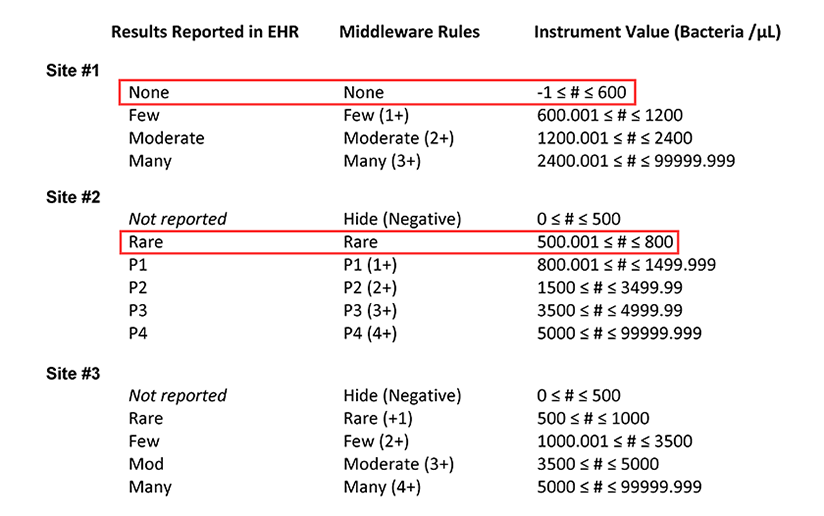 Table 1a. Summary of the middleware interpretation of the instrument values for number of bacteria per µL and the corresponding results reported in the electronic health record (EHR) for each site. This table highlights how a bacteria count of 600 /µL is reported at sites #1 and #2.
Table 1a. Summary of the middleware interpretation of the instrument values for number of bacteria per µL and the corresponding results reported in the electronic health record (EHR) for each site. This table highlights how a bacteria count of 600 /µL is reported at sites #1 and #2.
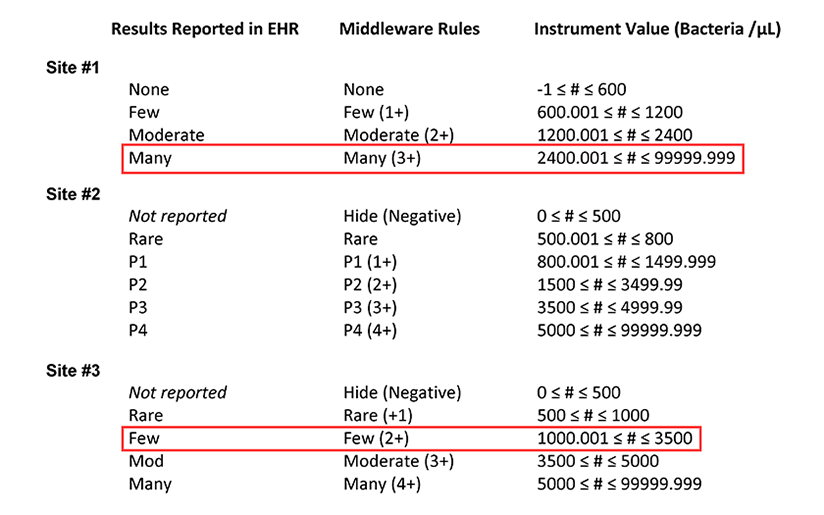 Table 1b. Summary of the middleware interpretation of the instrument values for number of bacteria per µL and the corresponding results reported in the electronic health record (EHR) for each site. This table highlights how a bacteria count of 3000 /µL is reported at sites #1 and #3.
Table 1b. Summary of the middleware interpretation of the instrument values for number of bacteria per µL and the corresponding results reported in the electronic health record (EHR) for each site. This table highlights how a bacteria count of 3000 /µL is reported at sites #1 and #3.
After looking at the tables provided by the resident, the laboratory supervisors are surprised to learn that if they ran the same sample at all three sites, different results could be reported in the EHR, with the potential to impact patient management. Furthermore, the team realizes that if a single reflex algorithm were implemented at all three sites, variations in the input parameters could mean that the same sample is subjected to urine culture at one laboratory but not at another.
You report to the hospital system’s infection control committee the challenge posed by the different practices at the three laboratories. You explain that you are working with the laboratory staff and vendor to first standardize the middleware rules and reporting in the EHR and provide a revised timeline to account for the extra steps that are necessary for setting up a standardized reflex testing protocol.
Middleware can optimize and improve the data flow between laboratory analyzers and the LIS. Among the many advantages of incorporating middleware into the laboratory includes the ability to create customizable rule sets for reflex testing and autoverification tasks.
Inclusion of the middleware rules in the SOP for the respective laboratory procedures so that they are subject to the same annual review process as the SOPs is a recommended practice. It is important to also evaluate whether the results are appearing correctly when transmitted to the EHR. Even when the same analyzer is used across laboratories, they may have different middleware rules which can significantly alter the reported results or downstream testing. It is also important to evaluate whether the raw data from analyzers is transmitted to the LIS for review, or if the only data received by the LIS is the middleware interpretation of the raw data.
When considering harmonizing data across disparate sites, for example, to establish a reflex algorithm, it is important to review the middleware rules applied to results from each analyzer. Variations in middleware rules may also have potential implications if the results are populating the same field in the EHR and will therefore look interchangeable to the patient facing colleagues. Finally, aggregation of data demands consistency of data type and format across sites.
- Middleware is frequently used by laboratories to connect laboratory analyzers with the LIS and may allow the laboratory to institute customizable rule sets for reflex testing and autoverification tasks.
- Middleware rules are often not immediately accessible by the laboratory staff and are not necessarily something that staff have familiarity with despite the critical role the rules play in the laboratory’s workflow; therefore, it is strongly recommended to include the middleware rules related to each laboratory procedure in their respective SOPs and to include the middleware rules in the annual SOP review process.
- When considering the implementation of any middleware software it is important to determine from the vendor if the raw data (original values generated by the analyzer), such as bacteria count in number /µL, will also be transmitted to the LIS, ensuring that the granular data is easily accessible and available to the laboratory as part of quality assurance/quality control review, or if the only data transmitted to the LIS will be the middleware’s interpretation of the results.
- It is the laboratory’s responsibility and a CAP checklist requirement that the laboratory verify that patient results are accurately transmitted by the laboratory into the patient’s records (i.e. the EHR). This verification must be performed prior to implementation of an interface (i.e. pre go-live), whenever any change is made to an existing interface that could affect the accuracy of transmission of patient results, and reverified at least every two years.
- When considering harmonizing data across disparate sites, for example, to establish a reflex algorithm, it is important to review the middleware rules applied to results from each analyzer at every site.
- Variations in middleware rules for a single test may lead to critically different interpretations and values that are not equivalent or interchangeable; it is important to consider and evaluate whether non-interchangeable/non-equivalent results will populate the same line in the EHR.
- If the middleware rules are setup to “hide” a negative result for bacteria in a urinalysis panel, based on the examples in this case, what result would you expect to see in the EHR?
- The EHR result for bacteria would be “Negative.”
- The EHR result for bacteria would be “Below the limit of detection.”
- The bacteria analyte and result would not be reported in the EHR.
- The EHR result for bacteria would be <600 bacteria /µL.
- Which is true about CAP checklist item GEN.43875 regarding requirements for validation of autoverification?
- Validation of the autoverification algorithm decision rules does not need to include specimens with analyte concentrations outside of the normal reference range.
- Validation of autoverification is only required once when the initial rules are set up.
- Validation of autoverification does not need to be performed annually but should be performed at least once every two years.
- Validation of autoverification should take place whenever there is a change to the system that could affect the autoverification logic.
- Which is true about the CAP checklist GEN. 48500 regarding requirements that results be accurately transmitted from the laboratory to patient reports?
- The laboratory director is only required to verify the accuracy of data transmission once when bringing on a new assay as part of the validation process.
- The laboratory director must verify the accuracy of data transmission annually.
- The laboratory director must verify the accuracy of data transmission at least every two years.
- The laboratory director is not responsible for verifying the accuracy of laboratory results transmitted to patient reports.
- The potential advantages of using middleware rules in the implementation of strategic diagnostic reflex algorithms, such as a urinalysis with reflex to urine culture protocol, include:
- simplifying the diagnostic process for clinicians.
- facilitating a laboratory’s implementation and automation of data and evidence-based protocols.
- reducing inappropriate laboratory utilization.
- all of the above.
Clinical and Laboratory Standards Institute (CLSI). Autoverification of Clinical Laboratory Test Results: Approved Guideline. CLSI document AUTO10-A (ISBN 1-56238-620-4). Clinical and Laboratory Standards Institute, 940 West Valley Road, Suite 1400, Wayne, Pennsylvania 19087-1898 USA, 2006.
Clinical and Laboratory Standards Institute (CLSI). Managing and Validating Laboratory Information System: Approved Guideline. CLSI document AUTO08-A. Clinical and Laboratory Standards Institute, Wayne, PA, 2006.
Cowan DF, et al. Validation of the laboratory information system. Arch Pathol Lab Med. 1998;122:239-244.
Department of Health and Human Services, Centers for Medicare & Medicaid Services. Clinical laboratory improvement amendments of 1988; final rule. Fed Register. 2003(Jan 24): [42CFR493.1291(a)].
Pantanowitz L. Pathology Informatics: Theory and Practice. American Society of Clinical Pathologists Press; 2012.
Perrotta PL, Karcher DS. Validating Laboratory Results in Electronic Health Records: A College of American Pathologists Q-Probes Study. Arch Pathol Lab Med. 2016;140(9):926-31.
Riben M. Laboratory Automation and Middleware. Surg Pathol Clin. 2015;8(2):175-86.
2019
Michelle Stram, MD, ScM, FCAP
CAP Informatics Committee
New York City Office of Chief Medical Examiner
Clinical Instructor, Department of Forensic Medicine
New York University
New York, NY
1. c
2. d
3. c
4. d