Clinical Summary
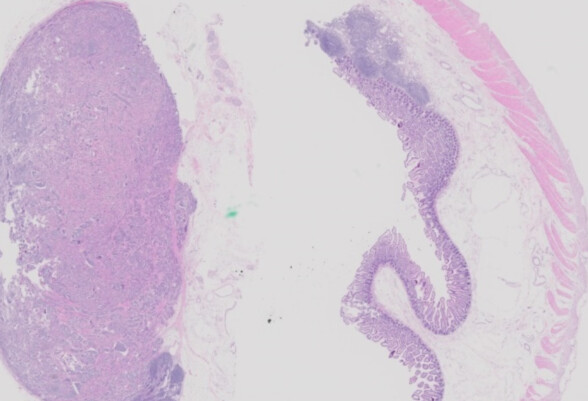
A 55-year-old man presents with a five-month history of abdominal pain, and a 10-pound weight loss. An abdominal computed tomography scan shows two 3-cm masses in the distal small bowel, along with enlarged mesenteric lymph nodes. Surgery is performed, and the gross specimen demonstrates two mural lesions and several enlarged lymph nodes. A section from the specimen is shown.
Master List of Diagnoses
- Epithelioid gastrointestinal stromal tumor
- Gangliocytic paraganglioma
- Metastatic melanoma
- Poorly differentiated neuroendocrine carcinoma
- Well-differentiated neuroendocrine tumor
Archive Case and Diagnosis
This case first appeared as Performance Improvement Program in Surgical Pathology (PIP) 2020, Case 08, and is well-differentiated neuroendocrine tumor in the ileum.
The information provided in this case was accurate and correct at the time of publication in 2020.
Any changes in terminology since the time of publication may not be reflected in this case.
Criteria for Diagnosis and Comments
Microscopic examination shows a well-differentiated lesion composed of islands, nests, cords, and rosettes of monotonous cells embedded in fibrous stroma. The cells have abundant amphophilic cytoplasm and round nuclei with stippled chromatin (a “salt-and-pepper” appearance). Some of the cells are enlarged, but mitotic figures are rare to absent, and there is no necrosis. Prominent retraction artifact surrounds the tumor nests, mimicking lymphovascular invasion (which is often also present). The neoplasm involves the small bowel wall, local lymph nodes, and the soft tissue of the mesentery. While the diagnosis can be suggested on H&E staining, immunohistochemical stains for pankeratin, synaptophysin, and chromogranin are positive in this case. Overall, the appearance is diagnostic for well-differentiated neuroendocrine tumor (WD-NET; formerly “carcinoid”) of the small intestine.
Midgut WD-NET of the jejunum/ileum has increased in incidence over the past few decades, and it is now the most common primary malignancy at this anatomic location, with adenocarcinoma coming in second. WD-NET have an incidence of approximately 1.0 case per 100,000 population in the United States. Patients are typically 50 to 70 years old, and the disease has no sex predilection. Small lesions are often asymptomatic, though larger or metastatic lesions can cause abdominal pain, intestinal obstruction, and gastrointestinal bleeding. Up to one-third of patients have multiple primary lesions, though the prognostic implication of multifocality appears minor at best. These neoplasms can secrete serotonin, which enters the hepatic portal system and is broken down by the liver. However, if the tumor metastasizes to the liver, then the serotonin can directly enter the systemic circulation and cause symptoms of “carcinoid syndrome” (flushing, diarrhea, wheezing, and/or heart disease).
Though WD-NET at this site are slow-growing, they should not be considered indolent. They can metastasize to local lymph nodes even when small, and liver metastasis may be seen at initial presentation. In addition to these typical sites of metastasis, WD-NET can also metastasize directly to mesenteric adipose tissue. Such mesenteric tumor deposits have recently been shown to harbor a worse prognosis than lymph node metastases, and they are now included as part of American Joint Committee on Cancer (AJCC) N-category staging in the latest (8th) edition. Although the overall 10-year survival is roughly 78%, patients may die of metastatic disease or of complications related to mesenteric disease and/or fibrosis compromising the mesenteric blood supply.
WD-NET of the jejunum/ileum appear fairly stable from a molecular perspective. The most common abnormality is loss of heterozygosity of chromosome 18. Occasional single nucleotide variants have been identified in a number of genes (including VHL, BRAF, and MEN1), and a small percentage of cases harbor inactivating insertions/deletions in CDKN1B. Still, there are currently no therapeutic options that target molecular characteristics of jejunal/ileal WD-NET. Instead, adjuvant therapeutic options for patients with metastatic disease include somatostatin analogues and interferon-α.
Naming and grading of neuroendocrine neoplasms continues to evolve. Per the most recent edition of the World Health Organization (WHO) Classification of Digestive System Tumours (from 2019), a neoplasm’s name should be determined by the H&E appearance: bland “carcinoid”-like lesions should be termed “well-differentiated neuroendocrine tumor,” and high-grade malignancies with nuclear atypia, necrosis and abundant mitoses should be termed “poorly differentiated neuroendocrine carcinoma.” (Equivocal intermediate cases are rare, especially in the gastrointestinal tract.)
Grading is performed by counting mitotic rate (per 10 high-power fields [hpfs]) and Ki-67 proliferative index by immunohistochemistry (per 500 to 2,000 cells). If a neoplasm has fewer than 2 mitoses per 10 hpfs and a Ki-67 rate less than 3%, it should be called grade 1. If it has more than 20 mitoses per 10 hpfs and/or a Ki-67 rate above 20%, it should be called grade 3. Tumors with parameters in between these cutoffs should be called grade 2. Poorly differentiated neuroendocrine carcinomas are essentially always grade 3 and actually are not assigned a grade in the new WHO system. Well-differentiated neuroendocrine tumors can be any grade; in the jejunum/ileum, most are grade 1, and grade 3 lesions are quite rare but do occur.
While WD-NET is generally straightforward to diagnose on H&E, a few other entities may enter the differential diagnosis. Poorly differentiated neuroendocrine carcinoma (PD-NEC) in the gastrointestinal tract resembles those in the lung and would show more atypia than WD-NET. Small-cell PD-NEC shows small, hyperchromatic, molding cells with minimal cytoplasm, and large-cell PD-NEC shows enlarged cells with ample cytoplasm but prominent nuclear atypia and nucleoli. Both typically show numerous mitotic figures and abundant necrosis. These high-grade lesions have a poor prognosis and are typically treated with platinum-based chemotherapy. WD-NET and PD-NEC are generally distinguished on H&E, as both can be grade 3 and both can express synaptophysin and chromogranin.
Gastrointestinal stromal tumor (GIST) is the most common mesenchymal neoplasm of the small intestine, though it occurs more commonly in the stomach. They are most commonly bland spindle-cell neoplasms with long, slender nuclei and abundant collagenous matrix. Less commonly, they may have epithelioid rather than spindled cells, and these examples are more likely to have a higher mitotic rate. While WD-NET very often metastasize to lymph nodes, this is exceedingly unusual in GIST. Both spindle-cell and epithelioid GISTs are positive for CD117 and DOG1 by immunohistochemistry, and negative for synaptophysin and chromogranin. Most GISTs harbor a KIT mutation; some instead have a PDGFRA mutation, and rare examples have other molecular defects.
Gangliocytic paraganglioma is a rare triphasic neoplasm practically unique to the duodenum/ampulla. It is slightly more common in males and may cause abdominal pain or gastrointestinal bleeding. Histologically, it is composed of plump epithelioid cells, bland spindled cells, and intermixed ganglion cells. Synaptophysin and chromogranin are positive in the epithelioid and ganglion cells, while S100 is positive in the ganglion and spindled cells. The epithelioid cells may also be positive for pankeratin. Fewer than 10% of cases metastasize to regional lymph nodes, and widely metastatic disease is extremely rare. It was recently discovered that this lesion may demonstrate a HIF2A gain-of-function mutation.
Melanoma is the most common lesion metastatic to the small intestine (and metastases as a whole are more common than primary small intestinal malignancies). Metastatic melanoma should show high-grade nuclear features, prominent nucleoli, and numerous mitoses; with luck, they may also show some amount of melanin pigment. Bland examples may mimic WD-NET by virtue of having several foci in the bowel wall and also involving regional lymph nodes. Immunohistochemistry can easily resolve this differential diagnosis, as melanoma is positive for S100, SOX10 and other melanoma markers, usually (approximately 70%) negative for synaptophysin and always negative for chromogranin.
Supplementary Questions
- A small bowel lesion shows a classic “carcinoid” appearance on H&E, 6 mitotic figures per 10 high-power fields, and a Ki-67 proliferative index of 25% by immunohistochemistry. What is the best diagnosis for this neoplasm?
- Poorly differentiated neuroendocrine carcinoma, large cell type, WHO grade 3
- Poorly differentiated neuroendocrine carcinoma, small cell type, WHO grade 3
- Well-differentiated neuroendocrine tumor, WHO grade 1
- Well-differentiated neuroendocrine tumor, WHO grade 2
- Well-differentiated neuroendocrine tumor, WHO grade 3
- Carcinoid syndrome occurs when hepatic metastases of a well-differentiated neuroendocrine tumor deposit which hormone directly into the systemic circulation?
- Gastrin
- Glucagon
- Insulin
- Serotonin
- Vasoactive intestinal peptide
- Which of the following immunohistochemical stains can help distinguish gangliocytic paraganglioma from well-differentiated neuroendocrine tumor?
- Chromogranin
- Ki-67
- Pancytokeratin
- S100
- Synaptophysin
References
- Amin MB, Gress DM, Meyer Vega LM, et al, eds. American Joint Committee on Cancer Staging Manual. 8th Edition. Springer: Switzerland, 2017; p. 375-388 and 523-530.
- Fata CR, Gonzalez RS, Liu E, Cates JM, Shi C. Mesenteric Tumor Deposits in Midgut Small Intestinal Neuroendocrine Tumors Are a Stronger Indicator Than Lymph Node Metastasis for Liver Metastasis and Poor Prognosis. Am J Surg Pathol. 2017;41(1):128-133.
- Klimstra DS, Klöppel G, La Rosa S, Rindi G. Classification of neuroendocrine neoplasms of the digestive system. In: WHO Classification of Tumours Editorial Board, eds. WHO Classification of Tumours: Digestive System Tumours. 4th Edition. Lyon: IARC, 2019; p.16-19.
- Oronsky B, Ma PC, Morgensztern D, Carter CA. Nothing But NET: A Review of Neuroendocrine Tumors and Carcinomas. Neoplasia. 2017;19(12):991-1002.
- Xavier S, Rosa B, Cotter J. Small bowel neuroendocrine tumors: From pathophysiology to clinical approach. World J Gastrointest Pathophysiol. 2016;7(1):117-124.
Answer Key
- Well-differentiated neuroendocrine tumor, WHO grade 3 (e)
- Serotonin (d)
- S100 (d)

