- Home
- Member Resources
- Articles
- CHIPping Away at CHIP (Or What It Means If Your Heme or Solid Tumor NGS Panel Identifies CH)
After reading the article, hear more background from the author on the CAPcast, CHIPping Away at CHIP.
Schtuff Happens as We Age
We all know it; we all live it. Aging wreaks all sorts of havoc with your body. Not surprisingly, those changes even occur at the DNA level. The normal aging process leads to the random acquisition of various “passenger” mutations in hematopoietic stem cells, approximately 14-20 mutations per year, predominantly in non-coding portions of the genome.2,3 When these mutations are identified in individuals with no evidence of a hematologic malignancy and normal peripheral blood counts, it is known as clonal hematopoiesis (CH).3-5 CH initially carried the moniker of CH of indeterminate potential when it was first identified in 2014, or CHIP, but after several years of intensive study, we now know that the potential isn’t quite as indeterminate as we once thought.
Incidence of CH

Figure 1 A. Incidence of CH (ref. 3), B. Incidence rate of all cancers (graphed from https://www.cancer.gov), and C. Incidence rate of MDS (graphed ref 10).
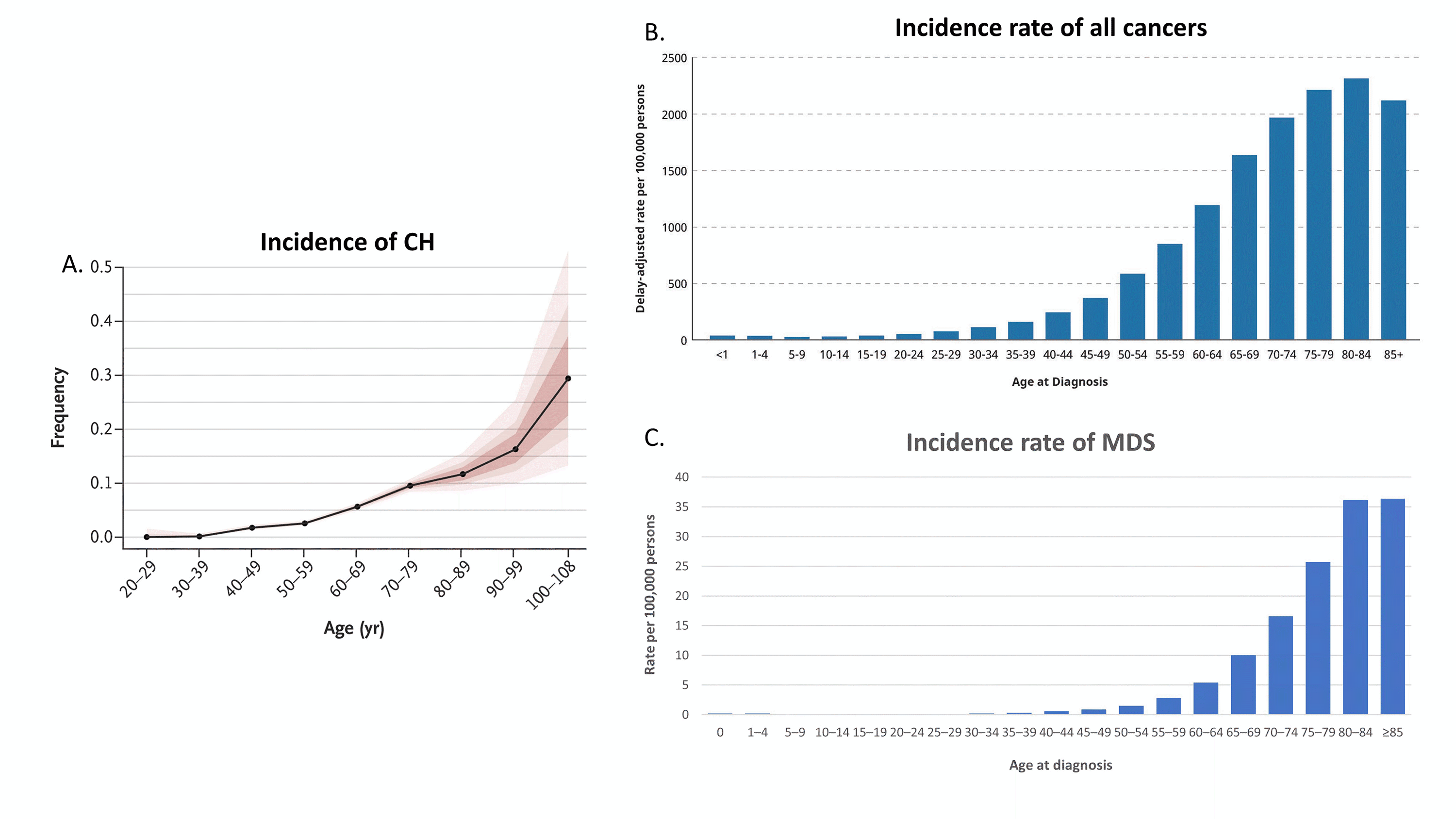
The incidence of CH rises rapidly after the age of 60, with 10-20% of individuals over the age of 70 with identifiable CH (Figure 1A). These initial studies examined the clinical implications of CH using a VAF cutoff of 2%, a level determined by the limit of detection of next generation sequencing (NGS) due to background PCR and sequencing noise. The use of error-corrected NGS, however, has identified CH, down to 0.03% VAF (median 0.24% VAF) in 95% of individuals 50-70 years of age.6 Indeed, the rise in incidence of any mutation(s) can be seen as early as age 30 even without error-corrected NGS, and some mutations, such as JAK2 p.V617F, have been identified even in utero.2-7 The incidence of CH parallels the rise in all cancers in general (Figure 1B-C).3-5, 8-11 Not surprisingly, it is common to see CH in the background of other malignancies, but CH has been identified in approximately 20-33% of cancer patients, significantly above the approximately 5-10% incidence expected from age matched controls.12-15 Thus, the enrichment of CH in cancer and the interplay of CH with these other neoplasms has garnered significant interest.
What Do You Mean that CH(ips) Aren’t Good for Your Health?

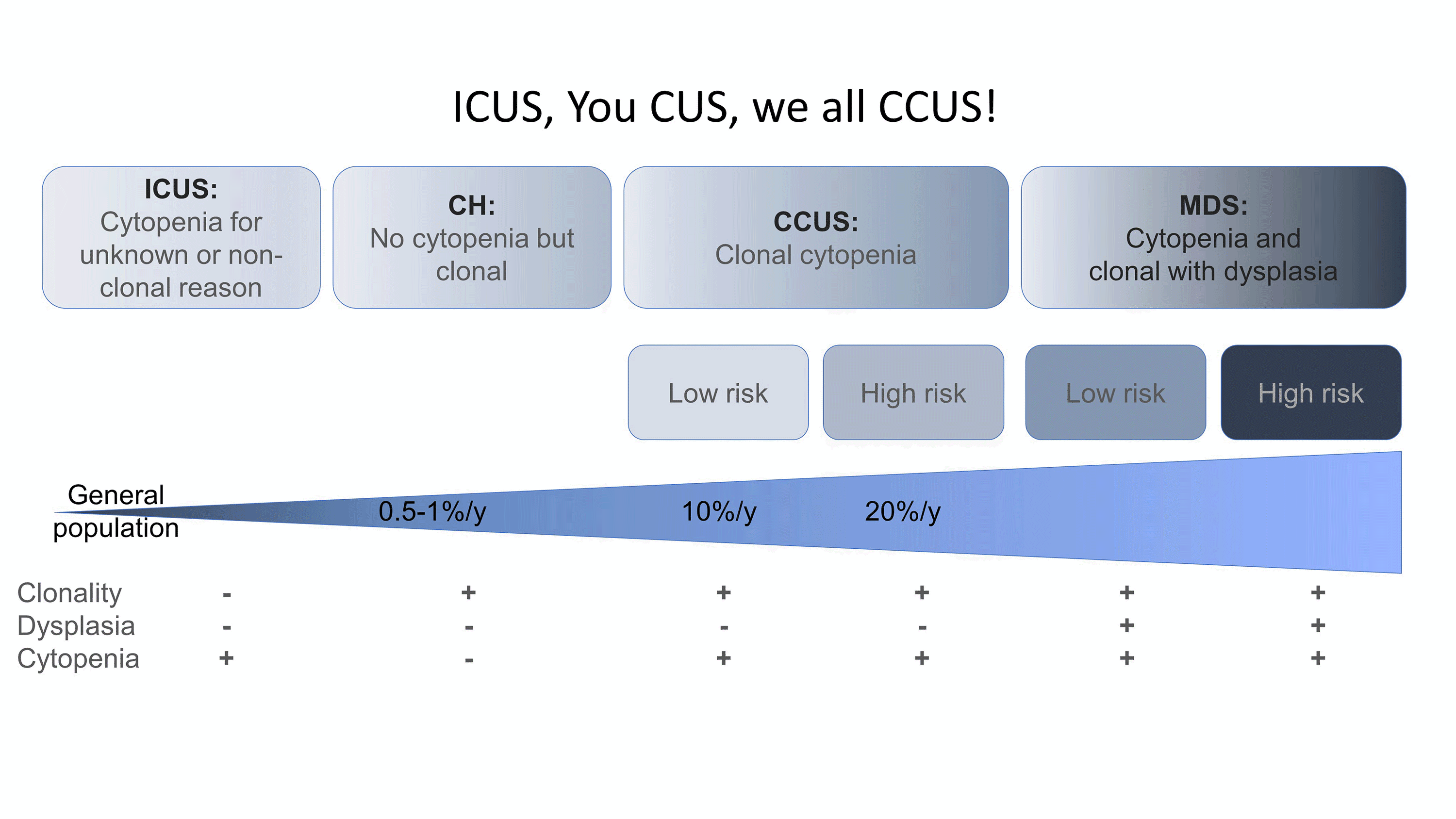
Figure 2. ICUS, You CUS, we all CCUS. Characteristics and the relative rates of progression/year of ICUS, CH, and CCUS to MDS (adapted from ref. 17 and 18)
Despite the initial tag of “indeterminate potential,” associations between CH and disease processes came to light quickly. The initial studies uncovered a striking association of CH with cardiovascular disease (CVD).16
Indeed, the presence of CH was a stronger predictor of coronary heart disease (hazard ratio 2.0) than more typical risk factors such as “body mass index” or “high blood pressure” with only “type II diabetes” and “age over 70” associated with a higher hazard ratio. In addition, CH also out-performed “high blood pressure” in risk of stroke (CH hazard ratio 2.4). Subsequent TET2 knockout mouse studies confirmed that decreases of functional TET2 resulted in excess accumulation of plaque in coronary arteries and the aorta in a dose dependent manner, illustrating the important role of various hematopoietic cells, such as neutrophils, monocytes, and platelets, in the etiology of atherosclerotic plaque. However, long-term studies are needed to determine if early intervention for CVD is beneficial in patients in whom there is identification of CH.
Although CH clones can remain stable for decades, CH has been associated with the development of subsequent hematopoietic neoplasms with a progression rate of approximately 0.5% to 1% per year, similar to that of other hematopoietic precursor lesions such as monoclonal B cell lymphocytosis or monoclonal gammopathy of uncertain significance.3,16 In the initial studies, the neoplasms were a mixture of both myeloid and lymphoid malignancies. However, the major concern is for myeloid neoplasms (MNs), with much of the focus on the spectrum of presentations between CH and myelodysplastic syndromes (MDS) (Figure 2). Patients may have CH without cytopenias (CH alone) or cytopenias without CH (idiopathic cytopenia of uncertain significance, ICUS). The combination of clonality and cytopenias results in clonal cytopenia of uncertain significance (CCUS), which carries variable risk of progression to a bona fide MDS characterized by cytopenias, clonality, and dysplasia.17-19 Indeed, the absence of pathogenic mutations by NGS in the peripheral blood (PB) of a cytopenic patient carries 95% NPV for the concurrent diagnosis of a MN in the bone marrow and accordingly PB NGS can be used as a comparatively inexpensive and minimally invasive screen to exclude patients who may not need a BM biopsy (Table 1).19,20 In patients with CCUS, a high-risk pattern is defined by 2 or more mutations, greater than 8.7% VAF, and particular genes and gene patterns, and arguably these patients should just be considered low risk MDS (Table 1).17
Table 1. Predictive value for a myeloid neoplasm based upon the number of mutations, variant allele fraction of mutations, and genes mutated in a selected cohort of patients with unexplained cytopenia and in an unselected cohort of patients with cytopenias. Abbreviations: VAF – variant allele fraction, PPV – positive predictive value, NPV – negative predictive value, Spliceosome – SF3B1, SRSF2, U2AF1, Epigenetic – TET2, ASXL1, DNMT3A, co-mutations – RUNX1, EZH2, CBL, BCOR, CUX1, TP53, IDH1/2.
|
|
Number pathogenic mutations |
VAF of mutations |
Genes mutated |
|
Selected cohort of patients with unexplained cytopenias (reference 17) |
Threshold |
>=1 |
8.70% |
Genes |
Spliceosome |
PPV |
0.81 |
0.86 |
PPV |
0.88-0.97 |
|
NPV |
0.76 |
0.77 |
Genes |
JAK2 |
|
Threshold |
>=2 |
10% |
PPV |
0.97 |
|
PPV |
0.88 |
0.86 |
Genes |
RUNX1 |
|
NPV |
0.58 |
0.77 |
PPV |
0.92 |
|
|
|
|
Genes |
Epigenetic |
|
|
|
|
PPV |
0.66-0.84 |
|
|
|
|
Genes |
Epigenetic + co-mutations |
|
|
|
|
PPV |
0.86-1.0 |
|
Unselected cohort of patients with cytopenias (reference 19) |
Threshold |
>=1 |
20% |
Genes |
Spliceosome |
PPV |
0.58 |
0.79 |
PPV |
0.67-1.0 (0.79 combined) |
|
NPV |
0.95 |
0.96 |
Genes |
Epigenetic |
|
Threshold |
>=2 |
|
PPV |
0.56-0.67 (0.55 combined) |
|
PPV |
0.75 |
|
Genes |
Signaling |
|
NPV |
0.88 |
|
PPV |
(0.67 combined) |
|
With error corrected NGS, CH can also be found in the lymphoid progeny of the affected hematopoietic stem cells.6 Indeed, cases of angioimmunoblastic T-cell lymphoma (AITL) harbor frequent mutations in the usual suspects TET2, DNMT3A, or IDH2.21-23 In these cases, identical mutations are found in the hematopoietic stem cell that subsequently can acquire additional hits and progress to either AITL and/or a MN. However, this particular clonal relationship between AITL and CH appears to be unique, and, in most cases of lymphoid neoplasms, CH is found in the myeloid compartment, distinct from the neoplastic lymphocytes.[unpublished data]
The Genes in CH: The Usual Suspects

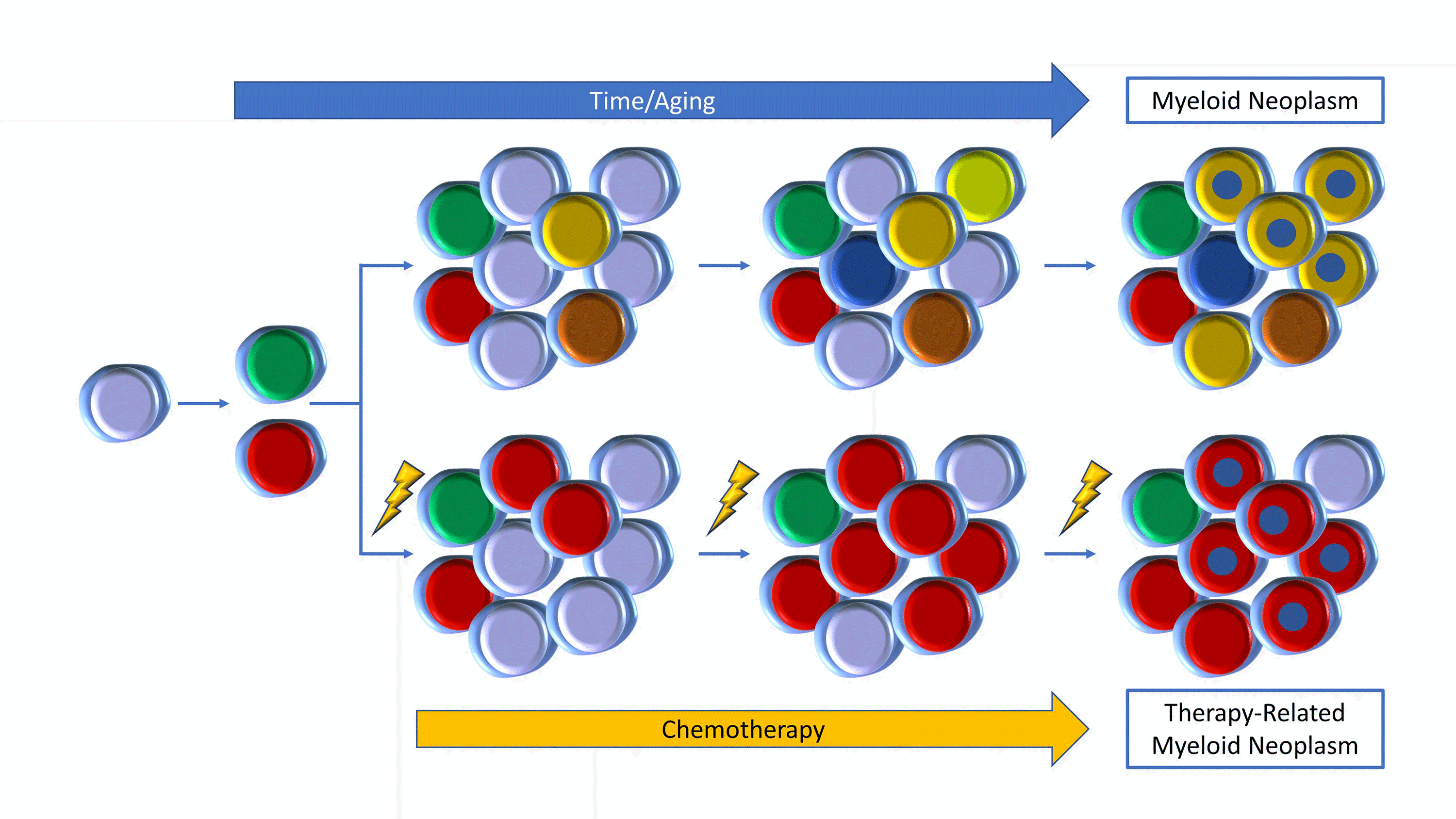
Figure 3. Top: Clones evolve with aging to development of a MN (yellow with blue). Bottom: Chemotherapy induces growth of DNA-damage gene mutations (red) to a t-MN (red with blue).
Although the majority of mutations acquired during the process of aging are non-coding and may represent passenger mutations,1 several genes are recurrently mutated in CH, fondly referred to as the Usual Suspects since they also can be altered across the spectrum of all MNs (Figure 3, top row). Epigenetic genes such as DNMT3A, ASXL1, and TET2, are the most common Usual Suspects, marking an aged stem cell, with DNMT3A alone accounting for approximately 50% of all CH mutations with TET2 and ASXL1 in more distant second and third place, respectively.3-5, 24 Older stem cells, such as found in individuals over the age of 70, are characterized by spliceosome mutations, such as SF3B1, and SRSF2.25 These 5 genes also happen to be the top 5 genes mutated in MDS, in which epigenetic and splicing gene mutations are recurrent founder driver mutations, strengthening the parallels between MDS and CH.26,27 Other frequent CH genes include JAK2, PPM1D, TP53, and some RAS pathway mutations such as CBL, GNAS, GNB1, NRAS, or KRAS.3,4,24
DNMT3A mutations play a unique role in CH, and not all DNMT3A variants are created equally. The DNTM3A hotspot, p.R882, makes up the majority of mutations in bona fide MNs, including approximately 60% of the mutations in this gene in AML.28 By contrast, DNMT3A p.R882 variants make up less than 20% of all DNMT3A mutations in CH [16.6% calculated from reference 3].3 Whereas the p.R882 variants demonstrate a strong dominant negative phenotype with only approximately 20% of normal DNA methylation function, other variants in DNMT3A demonstrate a spectrum of less dramatic hypomorphic phenotypes.29,30 Thus, while the p.R882 variants are enriched in MNs, the non-R882 variants predominate in CH. All DNMT3A hypomorphic variants appear to result in large undermethylated canyons (unmethylated regions over 3.5 kb) that result in higher expression of genes associated with increased stem cell self-renewal.24,31
Of note, CH can also manifest as mosaic chromosomal alterations (copy number variations) in addition to somatic mutations. These have been detected in patients who subsequently developed (therapy related) myeloid neoplasms and may represent an important, but less well understood component of CH that, in combination with somatic mutations, can result in biallelic targeting of certain genes such as TP53, TET2, and JAK2.32-34 These may be identified by chromosomal microarray as well as by some NGS panels. In addition, there are rare germline polymorphisms that may predispose the individual to developing CH, including variants in TERT, TET2, DNMT3A, and JAK2 and in the non-coding region of HAPLN1 enhancers.35
CH(IPS) Make You Fit or Unfit?
The superior fitness of the stem cell with CH has been invoked by several studies24,31 and explains the likelihood of various CH clones to expand under adverse bone marrow environments. Allogeneic stem cell transplantation (SCT) studies have demonstrated that CH confers a modest survival advantage in stem cells harboring these mutations.36 Forced to engraft in an adverse bone marrow environment caused by conditioning regimens, CH in the donor cells expanded overall 2.3-fold in the recipients compared to in the original donors, with the mean VAF achieved by the donor CH in the recipient approximately 10%. Donor marrow harboring CH has been associated with more rapid leukocyte engraftment.37 In addition, there is a potentially beneficial donor CH versus leukemia effect, especially if the donor-derived clone had a DNMT3A mutation. Conversely, there is significant adverse donor CH versus host effect leading to chronic graft versus host disease and the rare specter of donor-cell leukemia. Overall, there was no overt effect of donor-derived CH on non-relapse mortality or long-term survival, forestalling questions of screening SCT donors for CH.
The apparent expansion of CH under adverse conditions has also been shown to be true for autologous stem cell transplantation (ASCT) and, indeed, for any systemic chemotherapy or radiotherapy, supporting the association between CH and its sequelae with cancer therapy (Figure 3, bottom row). Cancer patients who have received certain types of chemotherapy (such as radiotherapy and certain cytotoxic agents, especially topoisomerase II inhibitors and carboplatin) have a risk of CH that rises with cumulative exposure to the therapies.38 In many cases, these clones were present at a lower level prior to any intervention, and expanded as a result of therapy.12,13,38 In particular, DNA damage repair genes, such as CHEK2, PPM1D, TP53, or ATM, are highly associated with prior therapy exposures,38 explaining the disproportionate increase in mutations in PPM1D in CH of cancer patients compared to the general population.13 Other cancer-predisposing exposures were also examined and smoking history was most associated with the mutations in ASXL1 with additional associations with SRSF2, SF3B1, and JAK2. Unlike in the setting of SCT in which there are mixed sequelae associated with CH, presence of CH in the cancer patient dramatically increases the occurrence of therapy-related myeloid neoplasms (t-MNs, either MDS or acute myeloid leukemia, AML). In lymphoma patients undergoing ASCT, the 10-year cumulative incidence of t-MN was 4.3% in patients without CH, but 14.1% in CH lymphoma patients.13 In breast cancer patients, the 10-year cumulative incidence of t-MN was to less than 1% in non-CH patients but increased to 9% in patients with CH.38 In many cases, the original CH clone expanded and acquired additional mutations, often involving TP53 or PPM1D. Occasionally, an initial CH clone might remain stable or even decrease in size while a second clone, potentially below the limit of detection initially, expanded in the t-MN.12,13,38
Robust Framework for Myeloid Mutational Patterns

Figure 4. Passengers, Drivers, Backseat Drivers, and Boston Drivers. Framework schematic of the sequential mutation acquisition seen in myeloid neoplasms.
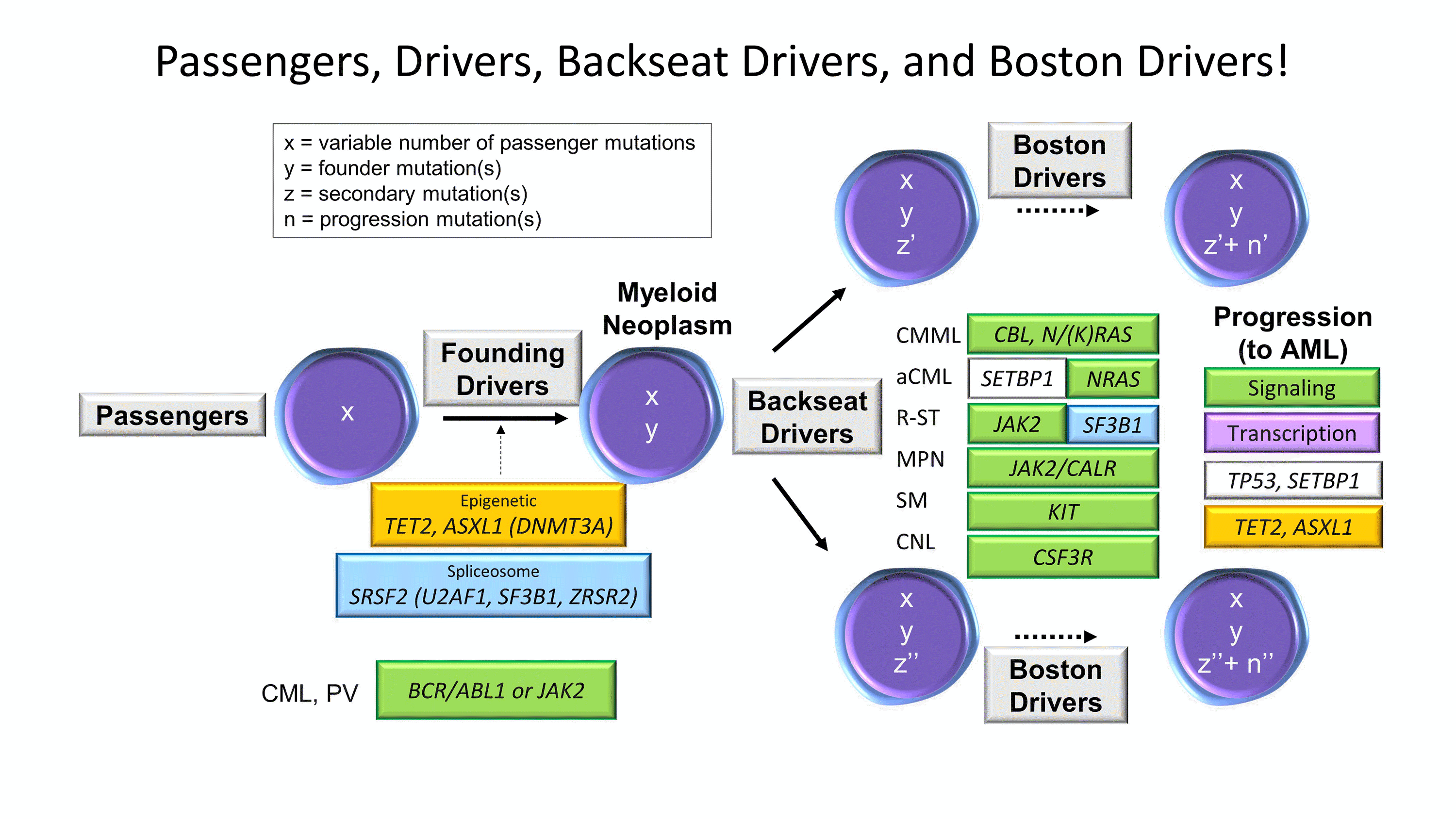
Whether de novo or therapy-derived, MNs encompass a diverse group of hematopoietic malignancies that originate from common myeloid progenitor stem cells with sequential accumulation of molecular genetic alterations to confer a survival and/or growth advantage. Recurring patterns of mutations in MNs are easily recognized and emphasize the importance of pathway disruption rather than individual mutations or genes (Figure 4). The initial founding drivers, on a background of passenger mutations, are often epigenetic and spliceosome pathway components- the same Usual Suspects found in CH. These can harbor numerous subclonal lesions, some of which may assume the role of telling the MN how to behave without actually driving the car, the backseat drivers, that may be enriched in specific disease entities. In some cases of myeloproliferative neoplasms (MPNs) or MDS/MPNs, these mutations are in the signal transduction pathway genes, such as JAK2, CALR, MPL, SH2B3, NFE2, or CSF3R in the MPNs and CBL mutations in some MDS/MPNs. Any of the chronic MNs (MDS, MDS/MPNs, or MPNs) can progress to an acute leukemia at which point they acquire the aggressive driver (jokingly referred to as the Boston driver) mutations, involving genes in cell signaling and DNA transcription pathways (Boston drivers known for being temperamentally dysregulated and having trouble signaling) as well as other known progression events such as TP53, SETBP1, or additional epigenetic gene mutation acquisition. Within this framework, many myeloid mutational patterns can be interpreted.26 It should be noted that the CH Usual Suspects are all clear founding drivers (predominantly epigenetic and splicing) and that the aged stem cell with CH serves as the foundation upon which most adult and/or therapy-related MNs are built.
Listen to the podcast
CHIPping Away at CHIP featuring Dr. Annette Kim, member of the CAP’s Personalized Healthcare Committee
References
- Welch JS, Ley TJ, Link DC, Miller CA, Larson DE, Koboldt DC, et al. The origin and evolution of mutations in acute myeloid leukemia. Cell. 2012 Jul 20;150(2):264–78.
- Williams N, Lee J, Moore L, Baxter J, Hewinson J, Dawson K, et al. Driver Mutation Acquisition in Utero and Childhood Followed By Lifelong Clonal Evolution Underlie Myeloproliferative Neoplasms. Blood. 2020;136(Supplment_2):LBA-1.
- Jaiswal S, Fontanillas P, Flannick J, Manning A, Grauman PV, Mar BG, et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med. 2014 Dec 25;371(26):2488–98.
- Genovese G, Kähler AK, Handsaker RE, Lindberg J, Rose SA, Bakhoum SF, et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med. 2014 Dec 25;371(26):2477–87.
- Xie M, Lu C, Wang J, McLellan MD, Johnson KJ, Wendl MC, et al. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat Med. 2014 Dec;20(12):1472–8.
- Young AL, Challen GA, Birmann BM, Druley TE. Clonal haematopoiesis harbouring AML-associated mutations is ubiquitous in healthy adults. Nat Commun. 2016 Aug 22;7:12484.
- Van Egeren D, Escabi J, Nguyen M, Liu S, Reilly CR, Patel S, et al. Reconstructing the Lineage Histories and Differentiation Trajectories of Individual Cancer Cells in Myeloproliferative Neoplasms. Cell Stem Cell. 2021 Mar 4;28(3):514-523.e9.
- Cogle CR. Incidence and Burden of the Myelodysplastic Syndromes. Curr Hematol Malig Rep. 2015 Sep 1;10(3):272–81.
- Cogle CR, Craig BM, Rollison DE, List AF. Incidence of the myelodysplastic syndromes using a novel claims-based algorithm: high number of uncaptured cases by cancer registries. Blood. 2011 Jun 30;117(26):7121–5.
- Ma X, Does M, Raza A, Mayne ST. Myelodysplastic syndromes: incidence and survival in the United States. Cancer. 2007 Apr 15;109(8):1536–42.
- Incidence rates by age at diagnosis, all cancer types. Source: SEER 21 2013–2017, all races, both sexes. [Internet]. Available from: https://www.cancer.gov/about-cancer/causes-prevention/risk/age Access date 06/17/2021
- Gillis NK, Ball M, Zhang Q, Ma Z, Zhao Y, Yoder SJ, et al. Clonal haemopoiesis and therapy-related myeloid malignancies in elderly patients: a proof-of-concept, case-control study. Lancet Oncol. 2017 Jan;18(1):112–21.
- Gibson CJ, Lindsley RC, Tchekmedyian V, Mar BG, Shi J, Jaiswal S, et al. Clonal Hematopoiesis Associated With Adverse Outcomes After Autologous Stem-Cell Transplantation for Lymphoma. J Clin Oncol Off J Am Soc Clin Oncol. 2017 May 10;35(14):1598–605.
- Coombs CC, Zehir A, Devlin SM, Kishtagari A, Syed A, Jonsson P, et al. Therapy-Related Clonal Hematopoiesis in Patients with Non-hematologic Cancers Is Common and Associated with Adverse Clinical Outcomes. Cell Stem Cell. 2017 Sep 7;21(3):374-382.e4.
- Olszewski AJ, Chorzalska AD, Kim AS, Quesenberry PJ, Lopresti ML, Fenton MA, et al. Clonal haematopoiesis of indeterminate potential among cancer survivors exposed to myelotoxic chemotherapy. Br J Haematol. 2019;186(3):e31-e35.
- Jaiswal S, Natarajan P, Silver AJ, Gibson CJ, Bick AG, Shvartz E, et al. Clonal Hematopoiesis and Risk of Atherosclerotic Cardiovascular Disease. N Engl J Med. 2017 Jul 13;377(2):111–21.
- Malcovati L, Gallì A, Travaglino E, Ambaglio I, Rizzo E, Molteni E, et al. Clinical significance of somatic mutation in unexplained blood cytopenia. Blood. 2017 Jun 22;129(25):3371–8.
- Steensma DP, Bejar R, Jaiswal S, Lindsley RC, Sekeres MA, Hasserjian RP, et al. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood. 2015 Jul 2;126(1):9–16.
- Shanmugam V, Parnes A, Kalyanaraman R, Morgan EA, Kim AS. Clinical utility of targeted next-generation sequencing-based screening of peripheral blood in the evaluation of cytopenias. Blood. 2019 Dec 12;134(24):2222–5.
- Lucas F, Michaels PD, Wang D, Kim AS. Mutational analysis of hematologic neoplasms in 164 paired peripheral blood and bone marrow samples by next-generation sequencing. Blood Adv. 2020 Sep 22;4(18):4362–5.
- Lewis NE, Petrova-Drus K, Huet S, Epstein-Peterson ZD, Gao Q, Sigler AE, et al. Clonal hematopoiesis in angioimmunoblastic T-cell lymphoma with divergent evolution to myeloid neoplasms. Blood Adv. 2020 May 26;4(10):2261–71.
- Tiacci E, Venanzi A, Ascani S, Marra A, Cardinali V, Martino G, et al. High-Risk Clonal Hematopoiesis as the Origin of AITL and NPM1-Mutated AML. N Engl J Med. 2018 Sep 6;379(10):981–4.
- Couronné L, Bastard C, Bernard OA. TET2 and DNMT3A mutations in human T-cell lymphoma. N Engl J Med. 2012 Jan 5;366(1):95–6.
- Challen GA, Goodell MA. Clonal hematopoiesis: mechanisms driving dominance of stem cell clones. Blood. 2020 Oct 1;136(14):1590–8.
- McKerrell T, Park N, Moreno T, Grove CS, Ponstingl H, Stephens J, et al. Leukemia-associated somatic mutations drive distinct patterns of age-related clonal hemopoiesis. Cell Rep. 2015 Mar 3;10(8):1239–45.
- McClure RF, Ewalt MD, Crow J, Temple-Smolkin RL, Pullambhatla M, Sargent R, et al. Clinical Significance of DNA Variants in Chronic Myeloid Neoplasms: A Report of the Association for Molecular Pathology. J Mol Diagn JMD. 2018 Nov;20(6):717–37.
- Mossner M, Jann J-C, Wittig J, Nolte F, Fey S, Nowak V, et al. Mutational hierarchies in myelodysplastic syndromes dynamically adapt and evolve upon therapy response and failure. Blood. 2016 01;128(9):1246–59.
- Ley TJ, Ding L, Walter MJ, McLellan MD, Lamprecht T, Larson DE, et al. DNMT3A mutations in acute myeloid leukemia. N Engl J Med. 2010 Dec 16;363(25):2424–33.
- Kim SJ, Zhao H, Hardikar S, Singh AK, Goodell MA, Chen T. A DNMT3A mutation common in AML exhibits dominant-negative effects in murine ES cells. Blood. 2013 Dec 12;122(25):4086–9.
- Russler-Germain DA, Spencer DH, Young MA, Lamprecht TL, Miller CA, Fulton R, et al. The R882H DNMT3A mutation associated with AML dominantly inhibits wild-type DNMT3A by blocking its ability to form active tetramers. Cancer Cell. 2014 Apr 14;25(4):442–54.
- Jeong M, Sun D, Luo M, Huang Y, Challen GA, Rodriguez B, et al. Large conserved domains of low DNA methylation maintained by Dnmt3a. Nat Genet. 2014 Jan;46(1):17–23.
- Loh P-R, Genovese G, Handsaker RE, Finucane HK, Reshef YA, Palamara PF, et al. Insights into clonal haematopoiesis from 8,342 mosaic chromosomal alterations. Nature. 2018 Jul;559(7714):350–5.
- Takahashi K, Wang F, Kantarjian H, Song X, Patel K, Neelapu S, et al. Copy number alterations detected as clonal hematopoiesis of indeterminate potential. Blood Adv. 2017 Jun 27;1(15):1031–6.
- Gao T, Ptashkin R, Bolton KL, Sirenko M, Fong C, Spitzer B, et al. Interplay between chromosomal alterations and gene mutations shapes the evolutionary trajectory of clonal hematopoiesis. Nat Commun. 2021 Jan 12;12(1):338.
- Bick AG, Weinstock JS, Nandakumar SK, Fulco CP, Bao EL, Zekavat SM, et al. Inherited causes of clonal haematopoiesis in 97,691 whole genomes. Nature. 2020 Oct;586(7831):763–8.
- Boettcher S, Wilk CM, Singer J, Beier F, Burcklen E, Beisel C, et al. Clonal hematopoiesis in donors and long-term survivors of related allogeneic hematopoietic stem cell transplantation. Blood. 2020 Apr 30;135(18):1548–59.
- Frick M, Chan W, Arends CM, Hablesreiter R, Halik A, Heuser M, et al. Role of Donor Clonal Hematopoiesis in Allogeneic Hematopoietic Stem-Cell Transplantation. J Clin Oncol Off J Am Soc Clin Oncol. 2019 Feb 10;37(5):375–85.
- Bolton KL, Ptashkin RN, Gao T, Braunstein L, Devlin SM, Kelly D, et al. Cancer therapy shapes the fitness landscape of clonal hematopoiesis. Nat Genet. 2020 Nov;52(11):1219–26.

Annette S. Kim, MD, PhD, FCAP, is the Henry Clay Bryant Professor and Division Head of Diagnostic Genetics and Genomics at the University of Michigan. Dr. Kim’s research program has focused on the study of hematolymphoid malignancies, including miRNAs in myelodysplastic syndromes, myeloid and lymphoid mutational patterns, and test utilization management. She has served as a member of Molecular Oncology Committee and is currently chair of the Personized Healthcare Committee for College of American Pathologists. She is also program chair of the Association for Molecular Pathology and past chair of the ASH Subcommittee on Precision Medicine. In 2019, Dr. Kim was awarded the CAP Public Service Award, and in 2025 she received the CAP Laboratory Improvement Programs Service Award.