- Home
- Member Resources
- Articles
- Immunohistochemical Surrogates of Molecular Genetic Alterations in Soft Tissue Tumors
Immunohistochemical (IHC) markers can serve as surrogates for molecular genetic events, providing a practical tool for early and accurate diagnosis. These markers are particularly useful in soft tissue tumor pathology, where both benign and malignant neoplasms often have defining genetic events that result in protein overexpression or loss (Table 1). Antibodies directed against protein products can be applied in daily practice and are particularly useful in diagnostically challenging specimens, such as small biopsies with scant tissue. The following are selected examples of soft tissue tumors with available surrogate IHC markers.
Table 1. Selected Immunohistochemical Surrogate Markers of Genetic Events in Mesenchymal Tumors
| Antigen | Tumor type(s) |
| ALK, ROS1 | Epithelioid inflammatory myofibroblastic sarcoma (ALK only), inflammatory myofibroblastic tumor |
| β-catenin | Desmoid fibromatosis, intranodal palisaded myofibroblastoma, pseudoendocrine sarcoma, sinonasal glomangiopericytoma |
| CADM3 | Superficial CD34-positive pleomorphic fibroblastic tumor |
| CAMTA1 | Epithelioid hemangioendothelioma |
| CYP1A1 | Angiofibroma of soft tissue |
| DDIT3 | Myxoid liposarcoma |
| DUX4, ETV4 | CIC-rearranged sarcoma |
| FOS, FOSB | Epithelioid hemangioma, osteoid osteoma, osteoblastoma, proliferative fasciitis/myositis, pseudomyogenic hemangioendothelioma (FOSB only) |
| FOXO1 | Alveolar rhabdomyosarcoma |
| GLI1 | GLI1-altered mesenchymal neoplasms |
| H3F3A G34W | Giant cell tumor of bone |
| H3F3B K36M | Chondroblastoma |
| H3K27me3 (loss) | Malignant peripheral nerve sheath tumor |
| INI1 (loss) | Epithelioid sarcoma, epithelioid malignant peripheral nerve sheath tumor, poorly differentiated chordoma |
| MDM2 | Well-differentiated and dedifferentiated liposarcomas |
| MUC4 | Low-grade fibromyxoid sarcoma, sclerosing epithelioid fibrosarcoma |
| NKX2.2 | Ewing sarcoma |
| NUTM1 | NUTM1-rearranged sarcoma |
| Pan-TRK | Infantile fibrosarcoma, NTRK-rearranged mesenchymal neoplasms |
| PDGFRA | PDGFRA-mutant gastrointestinal stromal tumor, inflammatory fibroid polyp |
| PRKAR1A (loss) | Cardiac myxoma, superficial angiomyxoma, malignant melanotic nerve sheath tumor |
| PLAG1 | Lipoblastoma |
| STAT6 | Solitary fibrous tumor |
| SSX, SS18::SSX | Synovial sarcoma |
Synovial Sarcoma
Among recently established surrogate markers are SSX (C-terminus) and SSX::SS18 for synovial sarcoma (SS). SS accounts for 5% to 10% of all soft tissue sarcomas and affects men and women equally, with a wide age range but a marked predilection for young adults.1 Approximately 70% of SS arise in the deep soft tissue of the lower extremities, followed by the trunk (15%), head and neck (7%), and less common locations like the retroperitoneum and thoracic cavity.2 SS exhibits aggressive behavior, with metastasis developing in up to 50% of cases, most commonly to the lungs.1 Morphologic variants include monophasic, biphasic, and poorly differentiated.2 The monophasic variant, which is the most common, has overlapping spindle cells arranged in dense sheets or intersecting fascicles.2 The spindle cells are uniform with scant cytoplasm, hyperchromatic nuclei, and inconspicuous nucleoli. The intervening stroma may exhibit myxomatous change, hyalinization, calcification, ossification, wiry collagen strands, and dilated, thin-walled, branching (hemangiopericytoma-like) vessels.1 The biphasic variant contains an epithelial component, with cells arranged in glands, nests, or cords.1 The epithelial cells are cuboidal to columnar, with ovoid vesicular nuclei and abundant pale eosinophilic cytoplasm. The poorly differentiated variant (which is associated with greater metastatic potential) is characterized by sheets of usually round (but occasionally spindled) tumor cells, greater nuclear atypia, and markedly increased mitotic activity.2
Conventional IHC markers often positive in SS include BCL2, CD99, EMA, and TLE1, although they are not specific.2 In tumors with an epithelial component, there is expression of various keratins, which can also be positive in up to 50% of poorly differentiated SS.2 Given the lack of specificity of such markers, most pathologists pursue fluorescence in situ hybridization or next-generation sequencing to confirm the diagnosis. Reviewing the published literature, Sreekantaiah and colleagues identified 38 of 42 published SS cases with a specific t(X;18)(p11.2;q11.2).3 Subsequent studies showed that t(X;18) results in the fusion of two genes, SS18 at 18q11 (previously designated SYT) and SSX at Xp11, with further studies revealing that the Xp11 breakpoint usually involves either of two closely related genes, SSX1 (Xp11.23) or SSX2 (Xp11.21).4 Baranov et al. later reported a novel, highly sensitive (95%) and specific (100%) SS18-SSX fusion-specific antibody and a second highly sensitive (100%) and specific (96%) SSX C-terminus antibody. Zaborowski et al. independently validated the utility of these antibodies. SSX and SS18::SSX are now routinely used in pathology practice (Figure 1).5
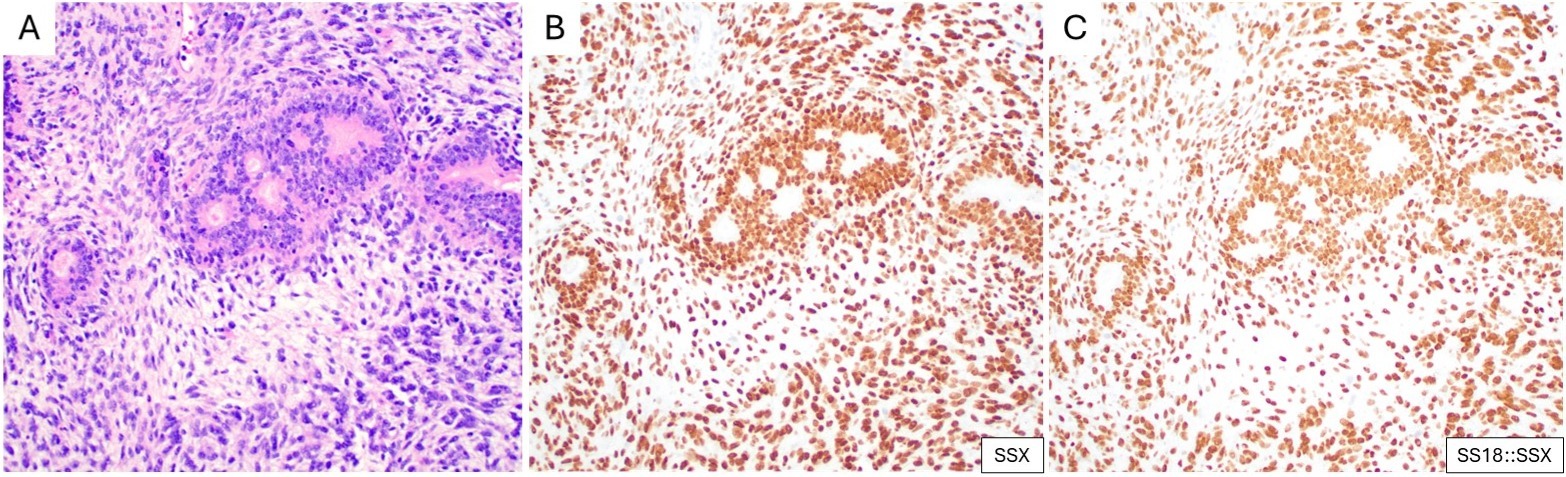
(A) Biphasic synovial sarcoma, with epithelial glandular structures admixed with uniform spindle cells (H&E). Corresponding IHC for (B) SSX and (C) SS18::SSX both show strong and diffuse nuclear expression in the two components.
Low-grade Fibromyxoid Sarcoma and Sclerosing Epithelioid Fibrosarcoma
Low-grade fibromyxoid sarcoma (LGFMS) and sclerosing epithelioid fibrosarcoma (SEF) are rare sarcomas within a morphologic spectrum.2 Histologically, LGFMS is characterized by sharply demarcated areas of alternating fibrous and myxoid areas, the latter with arcades of delicate blood vessels.6 The neoplastic cells are bland, short spindle cells with scant cytoplasm and elongated nuclei. A third of cases show “giant rosettes” composed of dense hyaline collagen peripherally encircled by epithelioid tumor cells.6 SEF is histologically defined by relatively bland epithelioid cells arranged in cords, strands, or nests, set within a densely hyalinized collagenous stroma.7 Molecularly, LGFMS harbors a FUS::CREB3L2 fusion in most cases, with FUS::CREB3L1 in a minority, and rarely EWSR1::CREB3L1; whereas SEF predominantly harbors EWSR1::CREB3L1, with FUS::CREB3L2 in a minority.8 Möller et al. sought to characterize LGFMS-specific gene expression patterns, and found that MUC4 was one of the top upregulated genes in LGFMS, with corresponding strong cytoplasmic immunohistochemical expression in 9 of 9 LGFMS cases.9 Doyle et al. examined a cohort of 49 LGFMS cases with FUS gene rearrangement and showed diffuse and intense cytoplasmic reactivity for MUC4 in all cases.10 The same group then studied MUC4 expression in SEF, finding diffuse and moderate to strong MUC4 expression in 78% of SEFs and 100% of tumors showing hybrid features of LGFMS and SEF.10 MUC4 expression did not predict the presence of FUS rearrangement (since EWSR1 rearrangements are more common in SEF).11 MUC4 is a dependable and easily applicable marker for the diagnosis of these two tumor types (Figure 2).
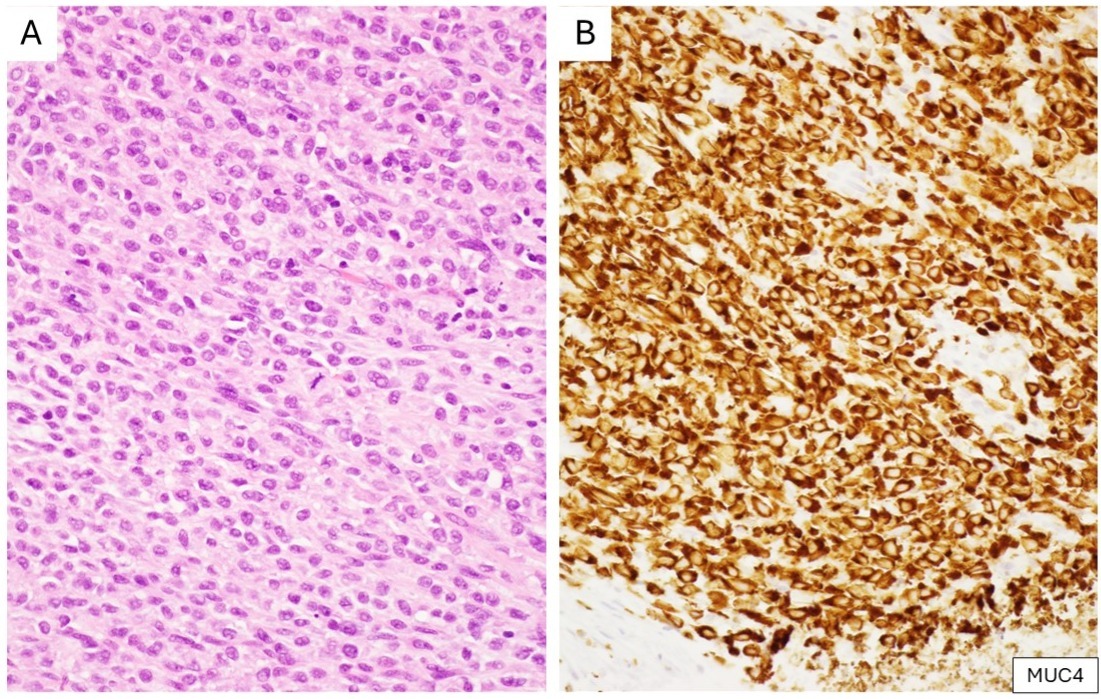
(A) Sclerosing epithelioid fibrosarcoma composed of epithelioid tumor cells within a dense collagenous stroma (H&E). (B) Corresponding IHC shows strong and diffuse cytoplasmic reactivity for MUC4.
Inflammatory Myofibroblastic Tumor and Epithelioid Inflammatory Myofibroblastic Sarcoma
Inflammatory myofibroblastic tumor (IMT) and epithelioid inflammatory myofibroblastic sarcoma (EIMS) are rare mesenchymal neoplasms, with distinct morphology and clinical behavior.2 IMT is a tumor of intermediate biological potential, mostly affecting children and young adults with a slight female predominance, and most commonly arising in the abdominopelvic region, lungs, and bladder.2,12 The prognosis is variable, with site-dependent recurrence rates ranging from 2% to 25% and a 2-3% risk of distant metastasis, most commonly to the lung and bones.12,13 Histologically, IMTs exhibit a broad morphological spectrum, ranging from hypocellular proliferations of spindled, ovoid, or stellate myofibroblasts with vesicular chromatin, small nucleoli, and pale eosinophilic cytoplasm, within a variable myxoid stroma with a prominent background inflammatory infiltrate of lymphocytes, plasma cells, and occasional eosinophils, to a densely cellular and fascicular or hypocellular, collagenous appearance.12,13
EIMS is considered a histologically and genetically distinct variant of IMT with an aggressive clinical course. There are approximately 43 cases currently reported in the literature.14,15 It typically arises in middle-aged to older adults, with a striking predilection for abdominopelvic sites. EIMS is notable for a pronounced tendency for local invasion, recurrence, and distant metastasis.14,15 Histologically, EIMS displays cytologically malignant epithelioid cells with vesicular chromatin large nucleoli, and amphophilic or eosinophilic cytoplasm, embedded within a myxoid commonly accompanied by a dense neutrophilic inflammatory infiltrate.15
First identified in IMT in the late 1990s, ALK gene rearrangements are frequent, with a wide range of fusion partners, including TPM3, TPM4, and CLTC.12,13,16 In EIMS, the most commonly reported ALK fusion partner is RANBP2.15 IHC directed against ALK quickly became a cornerstone in the diagnosis of IMT, with 50-60% of cases exhibiting cytoplasmic reactivity. ROS1 gene rearrangements have been identified in a subset of ALK-negative IMT, particularly in younger patients and lung-based lesions, with reported fusion partners including YWHAE and TFG.17 ROS1 IHC can be applied to identify IMTs with ROS1 gene rearrangement (Figure 3).18 By IHC, EIMS usually shows a distinctive nuclear membrane pattern of ALK (Figure 4).15 The identification of ALK and ROS1 overexpression by IHC (as surrogates for gene rearrangements) has important implications for targeted therapy, enabling the use of tyrosine kinase receptor inhibitors such as crizotinib in advanced or refractory cases.17
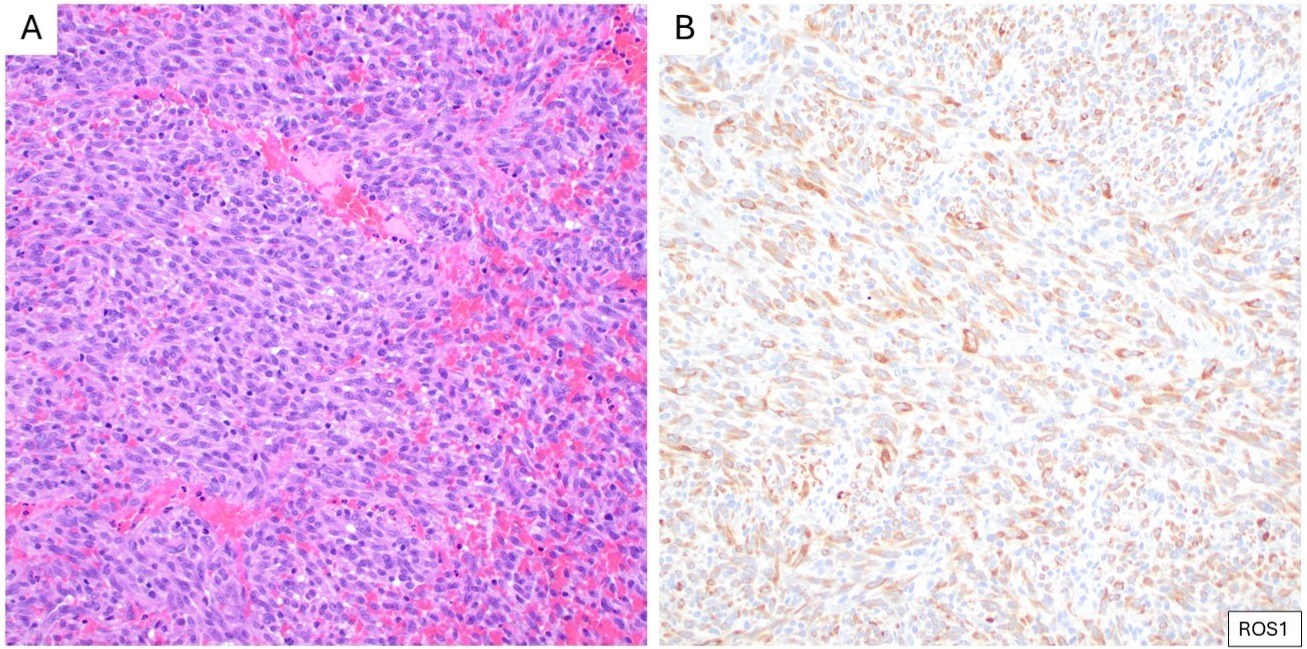
(A) Cellular inflammatory myofibroblastic tumor composed of densely packed spindle cells with mild atypia, arranged in a fascicular growth pattern with scattered lymphocytes (H&E). (B) Corresponding IHC demonstrates cytoplasmic expression of ROS1.
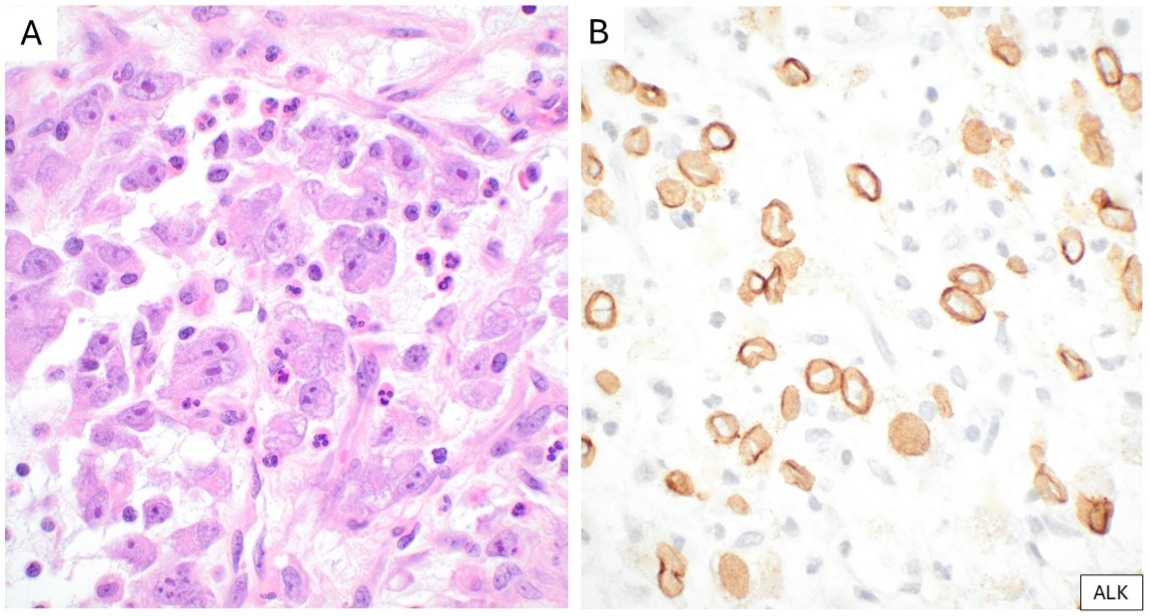
(A) Epithelioid inflammatory myofibroblastic sarcoma characterized by epithelioid tumor cells, some binucleated, with vesicular nuclei and large nucleoli, embedded in a myxoid stroma containing neutrophils. (B) Corresponding IHC strong nuclear membrane reactivity for ALK.
Myxoid Liposarcoma
First described by Evans in 1979, myxoid liposarcoma (MLPS) accounts for 20% to 35% of liposarcoma cases; this sarcoma type commonly arises in the lower extremities of adults in the third to fifth decades and is the most common liposarcoma in children and adolescents.2 As cellularity increases in MLPS, the risk of metastasis also rises; with an unusual metastatic predilection for bone, soft tissue, and retroperitoneum.2 Myxoid liposarcomas are typically hypocellular tumors with small uniform ovoid cells and uni- or bivacuolated lipoblasts within a prominent myxoid stroma containing a delicate “chicken wire” vascular pattern; atypia and mitotic activity are minimal.19 The tumor formerly known as round cell sarcoma is now recognized as part of the MLPS spectrum.19 It is characterized by hypercellular areas of undifferentiated round cells.19 Recognition of tumors with a predominant round cell component and minimal adipocytic differentiation can be challenging, particularly in limited biopsy specimens. Additionally, treated tumors can show decrease in cellularity, extensive stromal hyalinization, and maturation into white adipose tissue (cytodifferentiation). IHC for S100 is often positive but is not specific.19
MLPS is most often characterized by t(12;16)(q13;p11), resulting in FUS::DDIT3; up to 5% of cases instead harbor EWSR1::DDIT3.20 Utilizing a DDIT3 N-terminus mouse monoclonal antibody, Scapa et al. reported expression of DDIT3 by IHC in 100% of cases of MLPS, including high-grade MLPS and treated MLPS.21 Subsequently, Baranov et al. confirmed these results in high-grade MLPS, which was specific for MLPS among round cell sarcomas.22 The DDIT3 antibody is a therefore a surrogate marker for MLPS, most helpful in small biopsies and high-grade examples where the characteristic stroma and vascular patterns can be obscured (Figure 5).
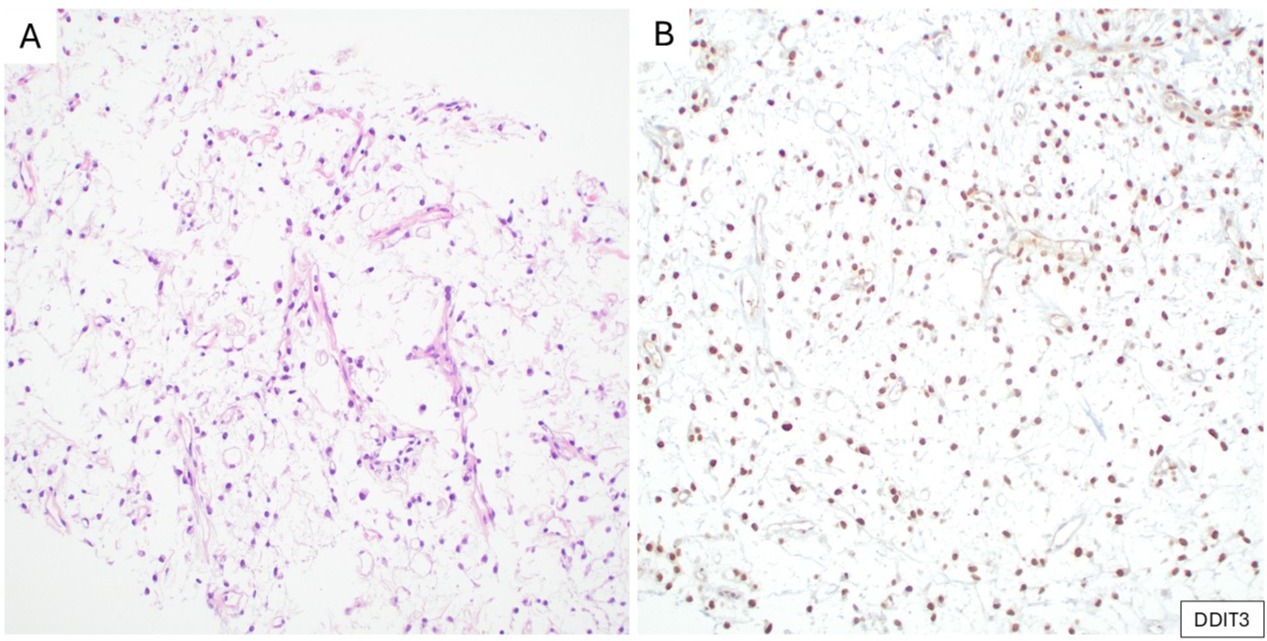
(A) Myxoid liposarcoma composed of uniform round to ovoid tumor cells and scattered univacuolated (signet-ring) lipoblasts, embedded in a myxoid matrix with a delicate, arborizing capillary vasculature (H&E). (B) Corresponding IHC demonstrates nuclear expression of DDIT3.
Epithelioid Hemangioendothelioma
Epithelioid hemangioendothelioma (EHE) is a malignant endothelial neoplasm with a slight female predominance, occurring in adults of any age, and most commonly arising in somatic soft tissue, lung, liver, or bone; visceral and osseous examples often present with multifocal disease.19 The clinical course of EHE is variable, ranging from indolent behavior in cutaneous tumors to more aggressive disease in soft tissue, where there is a 20% risk of metastasis.2
EHE is characterized by infiltrative cords, strands, or nests of epithelioid cells with round nuclei, small central nucleoli, and eosinophilic glassy cytoplasm, often set within a prominent myxohyaline stroma. Cytoplasmic vacuoles may be identified and serve as a helpful diagnostic clue. In up to 40% of cases, EHE is positive for keratins; EHE can therefore mimic metastatic carcinoma.19 If the diagnosis is suspected, IHC for vascular determinants, especially ERG and CD31, can be helpful.19 Other epithelioid endothelial neoplasms, namely epithelioid hemangioma and epithelioid angiosarcoma, can show significant histologic overlap.
A reciprocal t(1;3)(p36;q25) translocation, resulting in a WWTR1::CAMTA1 fusion, is characteristic of EHE.23 Shibuya et al. reported a CAMTA1 antibody with 87.5% sensitivity and 99.6% specificity for EHE, later validated by Doyle et al. (Figure 6). 24,25
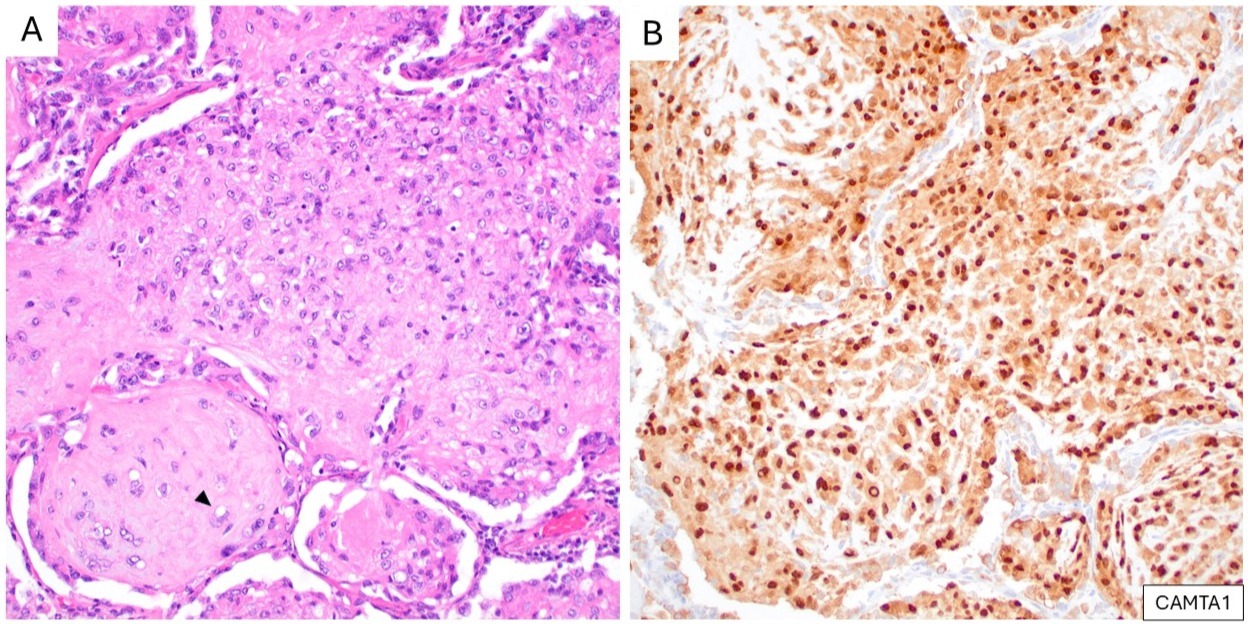
(A) A pulmonary metastasis of epithelioid hemangioendothelioma, showing epithelioid tumor cells with eosinophilic cytoplasm and intracytoplasmic vacuoles (arrowhead) (H&E). (B) Corresponding IHC demonstrates strong nuclear expression of CAMTA1.
Conclusion
Soft tissue pathology is a challenging field, primarily due to the rarity of tumors and overlapping histomorphology. IHC is a well-established and widely utilized technique; the availability of highly sensitive and specific surrogate markers for molecular genetic events improves diagnostic accuracy, reduces cost, and shortens turnaround time, ultimately enhancing patient management.
References
- Miettinen M, ed. Modern Soft Tissue Pathology: Tumors and Non-Neoplastic Conditions. Cambridge University Press; 2016.
- WHO Classification of Tumors Editorial Board. Soft Tissue and Bone Tumours. Vol. 3. 5th ed. Lyon: International Agency for Research on Cancer, 2020.
- Sreekantaiah C, Ladanyi M, Rodriguez E, Chaganti RS. Chromosomal aberrations in soft tissue tumors. Relevance to diagnosis, classification, and molecular mechanisms. Am J Pathol. 1994;144(6):1121-1134.
- Ladanyi M. Fusions of the SYT and SSX genes in synovial sarcoma. Oncogene. 2001;20(40):5755-5762.
- Zaborowski M, Vargas AC, Pulvers J, et al. When used together SS18-SSX fusion-specific and SSX C-terminus immunohistochemistry are highly specific and sensitive for the diagnosis of synovial sarcoma and can replace FISH or molecular testing in most cases. Histopathology. 2020;77(4):588-600.
- Evans HL. Low-grade fibromyxoid sarcoma: a clinicopathologic study of 33 cases with long-term follow-up. Am J Surg Pathol. 2011;35(10):1450-1462.
- Warmke LM, Yu W, Meis JM. Sclerosing Epithelioid Fibrosarcoma. Surg Pathol Clin. 2024;17(1):119-139.
- Matsuyama A, Hisaoka M, Shimajiri S, et al. Molecular detection of FUS-CREB3L2 fusion transcripts in low-grade fibromyxoid sarcoma using formalin-fixed, paraffin-embedded tissue specimens. Am J Surg Pathol. 2006;30(9):1077-1084.
- Möller E, Hornick JL, Magnusson L, Veerla S, Domanski HA, Mertens F. FUS-CREB3L2/L1-positive sarcomas show a specific gene expression profile with upregulation of CD24 and FOXL1. Clin Cancer Res. 2011;17(9):2646-2656.
- Doyle LA, Möller E, Dal Cin P, Fletcher CDM, Mertens F, Hornick JL. MUC4 is a highly sensitive and specific marker for low-grade fibromyxoid sarcoma. Am J Surg Pathol. 2011;35(5):733-741.
- Doyle LA, Wang W-L, Dal Cin P, et al. MUC4 is a sensitive and extremely useful marker for sclerosing epithelioid fibrosarcoma: association with FUS gene rearrangement. Am J Surg Pathol. 2012;36(10):1444-1451.
- Gleason BC, Hornick JL. Inflammatory myofibroblastic tumours: where are we now? J Clin Pathol. 2008;61(4):428-437.
- Coffin CM, Hornick JL, Fletcher CDM. Inflammatory myofibroblastic tumor: comparison of clinicopathologic, histologic, and immunohistochemical features including ALK expression in atypical and aggressive cases. Am J Surg Pathol. 2007;31(4):509-520.
- Chopra S, Maloney N, Wang WL. Epithelioid inflammatory myofibroblastic sarcoma with VCL-ALK fusion of central nervous system: case report and brief review of the literature. Brain Tumor Pathol. 2022;39(1):35-42.
- Mariño-Enríquez A, Wang W-L, Roy A, et al. Epithelioid inflammatory myofibroblastic sarcoma: An aggressive intra-abdominal variant of inflammatory myofibroblastic tumor with nuclear membrane or perinuclear ALK. Am J Surg Pathol. 2011;35(1):135-144.
- Lee C-J, Schöffski P, Modave E, et al. Comprehensive molecular analysis of inflammatory myofibroblastic tumors reveals diverse genomic landscape and potential predictive markers for response to crizotinib. Clin Cancer Res. 2021;27(24):6737-6748.
- Lovly CM, Gupta A, Lipson D, et al. Inflammatory myofibroblastic tumors harbor multiple potentially actionable kinase fusions. Cancer Discov. 2014;4(8):889-895.
- Hornick JL, Sholl LM, Dal Cin P, Childress MA, Lovly CM. Expression of ROS1 predicts ROS1 gene rearrangement in inflammatory myofibroblastic tumors. Mod Pathol. 2015;28(5):732-739.
- Hornick JL. Practical Soft Tissue Pathology: A Diagnostic Approach. 2nd ed. Elsevier; 2019.
- Crozat A, Aman P, Mandahl N, Ron D. Fusion of CHOP to a novel RNA-binding protein in human myxoid liposarcoma. Nature. 1993;363(6430):640-644.
- Scapa JV, Cloutier JM, Raghavan SS, Peters-Schulze G, Varma S, Charville GW. DDIT3 immunohistochemistry is a useful tool for the diagnosis of myxoid liposarcoma. Am J Surg Pathol. 2021;45(2):230-239.
- Baranov E, Black MA, Fletcher CDM, Charville GW, Hornick JL. Nuclear expression of DDIT3 distinguishes high-grade myxoid liposarcoma from other round cell sarcomas. Mod Pathol. 2021;34(7):1367-1372.
- Errani C, Zhang L, Sung YS, et al. A novel WWTR1-CAMTA1 gene fusion is a consistent abnormality in epithelioid hemangioendothelioma of different anatomic sites. Genes Chromosomes Cancer. 2011;50(8):644-653.
- Shibuya R, Matsuyama A, Shiba E, Harada H, Yabuki K, Hisaoka M. CAMTA1 is a useful immunohistochemical marker for diagnosing epithelioid haemangioendothelioma. Histopathology. 2015;67(6):827-835.
- Doyle LA, Fletcher CDM, Hornick JL. Nuclear expression of CAMTA1 distinguishes epithelioid hemangioendothelioma from histologic mimics. Am J Surg Pathol. 2016;40(1):94-102.

Maria Del Carmen Rodriguez Pena, MD, is a surgical pathology fellow with emphasis on soft tissue tumors at Brigham and Women’s Hospital, and she will continue subspecialty fellowship training in genitourinary pathology at Memorial Sloan Kettering Cancer Center. She completed her anatomic pathology residency at the NCI / NIH. Her interests center on the integration of morphologic, immunohistochemical, and molecular features in the classification of mesenchymal and genitourinary tumors.

Jason L. Hornick, MD, PhD, is the director of surgical pathology and immunohistochemistry and chief of soft tissue and bone pathology at Brigham and Women’s Hospital, professor of pathology at Harvard Medical School, and a consultant at the Dana-Farber Cancer Institute. Dr. Hornick’s research focuses on the development of diagnostic biomarkers for soft tissue tumors that correlate with molecular genetic alterations, which can be evaluated by conventional immunohistochemistry, and defining diagnostic criteria and clinicopathologic features of soft tissue tumors.