- Home
- Member Resources
- Articles
- Clonal Hematopoiesis and Inflammation
Clonal hematopoiesis of indeterminate potential (CHIP) refers to a clonal population of cells derived from a hematopoietic stem cell population with myeloid cancer-associated somatic mutations in an asymptomatic individual, present at a variant allele frequency ≥2%. The fundamentals of CHIP have been previously covered in an article on this page (CHIPping Away at CHIP (Or What It Means If Your Heme or Solid Tumor NGS Panel Identifies CH). CHIP affects more than 10% of healthy adults older than 65 years, whereas, when a variant allele frequency (VAF) of 0.03% is adopted, clonal hematopoiesis (CH) has been identified in 95% of individuals aged 50 to 70 years1,2. In addition to its prominent relationship with hematologic malignancies, emerging evidence has also associated CHIP with cardiovascular disease and inflammatory states. Risk factors for CHIP and cardiovascular disease and other inflammatory states appear in some instances to form a positive feedback loop, with significant overlap identified both on the genomic landscape of these disease processes and their phenotypical correlates. Over 80% of CHIP cases show DNMT3A, TET2 or ASXL1 driver gene mutations3.
Functions of common genes in CHIP
DNMT3A
DNMT3A promotes de novo DNA methylation of cytosine at CpG sites through its function as a DNA methyltransferase and regulates gene expression through epigenetic changes. DNMT3A is essential for embryonic differentiation. DNMT3A-null hematopoietic stem cells in murine models show differential methylation of loci with both increased and decreased methylation patterns, resulting in upregulation of multipotency genes and downregulation of differentiation factors.4,5 In malignancy, DNMT3A acts as a tumor suppressor, and somatic mutations in DNMT3A are all hypomorphic or loss-of-function, which favor self-renewal and promote expression of multipotency genes when present in hematopoietic stem cells, providing a competitive advantage for the clone4,6,7. Interestingly, DNMT3A variants involving p.R882, a mutational hotspot, also show dominant negative and gain of function activity. These variants are associated with higher leukemogenic potential, which may relate to reduced methyltransferase activity8.
TET2
TET2 also plays a key role in regulating DNA methylation and gene expression. TET2 encodes an iron and a-ketoglutarate dependent dioxygenase that initiates the demethylation sequence. TET2 oxidizes the methyl group added by DNMT3A by hydrolyzing 5'-methylcytosine (5'-mc) to 5'-hydroxymethylcytosine9; thus antagonizing methylation by DNMT enzymes. TET2 expression is also tied into histone modifications, the other major mechanism of epigenetic regulation of gene expression. Histone regulation includes a range of modifications, including methylation and demethylation of lysine residues by enzymes such as the Jumanji-C containing lysine demethylase 2A (KDM2A), with alteration in histone association with DNA affecting TET2 expression and subsequent DNA methylation patterns10. This complex interplay occurs in both normal and tumor cells where alterations in methylation status influence cellular differentiation and transformation11,12.
Epigenetic alterations in gene expression also affect inflammation by altering function of both lymphoid and myeloid inflammatory cells. TET2 expression inhibits inflammatory response in murine macrophages. TET2 expression also regulates Th1 and Th17 lymphocyte differentiation and cytokine expression, as well as Type 1 regulatory T cells. In the absence of Tet2, expression of IFN-gamma and IL-10 by the latter is inhibited13. Loss-of-function mutations are associated with increased production of IL-6 and expansion of the mutant clone14,15 Fuster et al. found in mouse models that partial reconstitution of the bone marrow with a TET2-deficient clone caused clonal expansion and accelerated atherosclerosis, thus linking CHIP expansion with atherosclerotic disease.
ASXL1
Like DNMT3A and TET2, ASXL1 regulates gene expression through epigenetic changes. In contrast to DNMT3A and TET2 which target DNA methylation, ASXL1 is involved in histone modifications. ASXL1 interacts with the polycomb repressive complex 2 (PRC2) with loss of ASXL1 resulting in reduced recruitment of PRC2 to specific targets and reduced histone H3 lysine 4 trimethylation (H3K27me3). ASXL1 is one of the most frequently mutated genes in myeloid neoplasms with similar mutations in CHIP16. Pathogenic variants of ASXL1 result in a truncated protein due to frameshift or nonsense mutations in exons 11 or 12 with loss of the C-terminal PHD domain, which is associated with histone interaction capacity. This truncated protein demonstrates altered function, including potential interaction with BET family proteins with bromodomains, including BRD4, which reads lysine methylation. The abnormal interaction results in gain of function and promotes activation of genes involved in myeloid malignancies17–19.
Inflammatory states that increase the risk of CHIP
Inflammation may contribute to expansion of CHIP clones or the progression of CHIP into malignancy20,21. Inflammatory20 and nutritional factors, such as obesity, smoking or availability of vitamin C, may potentially affect CHIP detection and clone size22,23.
Aging
Aging and inflammation are strongly correlated with cardiovascular events, and both show links with CHIP3,24. Aging is associated with the accumulation of mutations in hematopoietic stem cells, including an observed increase in the proportion of CHIP carriers and the size of clonal populations25,26. The relative fitness of acquired mutations in hematopoietic stem cells is modulated by environmental factors which include the pro-inflammatory microenvironment in aging26,27. Telomere length is also associated with CHIP, as evidenced by the increased prevalence of the latter in inherited telomeropathy cases28. In particular, germline variants, including in TERT29, have been associated with increased risk of certain CHIP mutations.
Diabetes and Obesity
Obesity is also recognized as a chronic inflammatory state, biased toward a Th1 response, and is associated with increased risk of type 2 diabetes mellitus, which may develop in part due to an increased inflammatory state30,31.Type 2 diabetes mellitus has been shown to mildly increase the risk of carrying CH-related mutations32. Obesity is also independently associated with CHIP, with normal weight or overweight body mass index status showing lower odds for CHIP than obese patients in a cohort of postmenopausal women23. In addition to obesity, chronically increased cholesterol levels from nutritional or other causes promote the proliferation and mobilization of hematopoietic stem cells and myeloid cell expansion, linking clonal hematopoiesis (as discussed further below)33,34.
Smoking
Smoking creates chronic lung inflammation primarily with preferential Th2 priming and results in upregulation of systemic inflammatory markers35,36. Smoking has been associated with number of CHIP mutations, most significant of which is the presence of ASXL1 mutations26.
Chronic inflammatory states
Chronic inflammatory conditions have been recognized as one of the elements modulating CHIP mutations20. For example, the frequency of DNMT3A mutations is higher in patients with ulcerative colitis21. However, they appear to show a reduced incidence of TET2-mutated CHIP clones, possibly due to an inflammatory microenvironment reducing the fitness of the TET2-mutated cells. However, the association of CHIP with autoimmune conditions is not universal as Savola et al. did not observe a significantly increased prevalence of CHIP in a population of patients with rheumatoid arthritis37.
VEXAS Syndrome
VEXAS syndrome (Vacuoles, E1 enzyme, X-linked, autoinflammatory, somatic) is a recently described syndrome caused by somatic mutations in UBA1 and shows an overlap of rheumatologic and hematological repercussions, including systemic inflammation, cytopenias and concurrent myelodysplastic syndromes in a subset38. Patients with VEXAS syndrome have a higher frequency of clonal hematopoiesis than similarly aged, older healthy adults. The most commonly involved genes in clonal hematopoiesis in these patients are DNMT3A and TET2. Gutierrez-Rodriguez posited that these mutations might confer a survival advantage in a highly-inflammatory microenvironment, as is found in VEXAS syndrome39,40. VEXAS may thus result in a vicious circle, with CHIP clones contributing to worsened inflammatory symptoms that in turn further stimulate CHIP expansion.
Chemotherapy agents
Cancer therapy is a source of selective pressure for hematopoietic stem cell clones, and different therapies may select for different mutant clones. Cancer therapy with radiation, platinum and topoisomerase II inhibitors preferentially selects for mutations in TP53, PPM1D and CHEK2 genes41.
Conditions where CHIP increases the risk for disease/inflammation

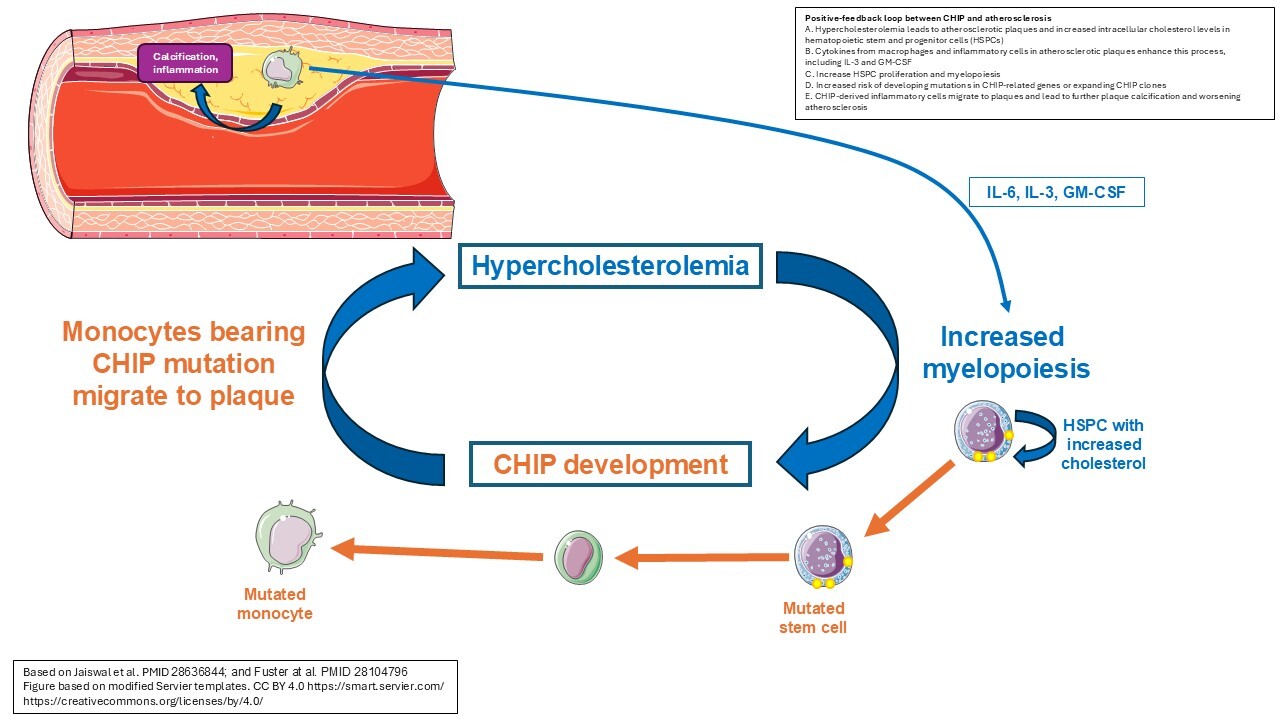
Atherosclerosis and elevated cholesterol
Heyde et al. referred to the interplay between hyperlipidemia, atherosclerotic plaque formation and chronic inflammation as the "atherosclerosis trait complex." These authors demonstrate that the rate of hematopoietic stem cell division is increased in both humans and mice with atherosclerosis, and that this elevated proliferation correlated with the development of clones with driver mutations for CHIP34. Altered cholesterol levels can directly impact the rate of proliferation of hematopoietic stem cells, with high HDL levels suppressing their proliferation. Conversely, increased cellular cholesterol levels due to deficient efflux leads to an increased proliferative responsiveness to IL-3 and GM-CSF, thus stimulating myelopoiesis42 (Figure 1). The production of cytokines by the inflammatory cells in atherosclerotic plaques may further enhance this process43. Jaiswal et al. found that mutations in any of the genes DNMT3A, TET2, ASXL1, or JAK2 were associated with coronary heart disease, and that individuals with clonal hematopoiesis also had increased coronary artery calcification, compounding the atherosclerotic burden33. These findings combined indicate a likely bidirectional relationship where atherosclerosis causes or enhances the process of clonal hematopoiesis through elevation of proliferation rates, and in turn CH further drives the atherosclerotic process34.
Cardiovascular disease
The prior article on this page also covers the intriguing relationship between CHIP and cardiovascular disease. To briefly summarize, Jaiswal et al. found an increased incidence of both coronary heart disease and ischemic stroke among individuals with clonal hematopoiesis with an identified somatic mutation32. This includes the association of the presence of common somatic mutations TET2, DNMT3A and ASXL1. Other mutations found in hematologic malignancies such as JAK2, which has a known relationship with thrombosis, were also associated with increased cardiovascular risk. In addition, CHIP mutations have also been associated with adverse outcomes in cardiovascular events such as an increase in coronary artery calcification, a 4-fold higher risk of having a myocardial infarction33,44, 2-fold risk in overall coronary heart disease and a 2.6-fold risk in developing ischemic stroke32.
Heart failure
Dorsheimer et al. found that the presence of DNMT3A or TET2 mutations correlated with worse outcomes (death and heart failure hospitalization) for patients with congestive heart failure (CHF) of ischemic origin, at a hazard ratio of 2.1 (p 0.02)44. They also detected a dose-response association between VAF and clinical outcome, suggesting a causal relationship between DNMT3A and TET2 CHIP mutations and the clinical outcome in patients with CHF.
Coronary heart disease
Jaiswal et al found that individuals from two cohorts with clonal hematopoiesis had a 1.9-fold greater risk of incident coronary heart disease (p=0.0002)33, defined as having myocardial infarction or coronary revascularization procedures. The authors also found a correlation between CHIP mutations and early-onset myocardial infarction. On a large cohort study, Bick et al. found a hazard ratio of 1.27 (p 0.019) for the correlation of CHIP with increased risk for cardiovascular events (defined here as a combination of myocardial infarction, coronary artery revascularization procedures, stroke or death)45. When stratified by size of the clone based on VAF, large CHIP clones (VAF >10%) conferred the most risk, at 1.50-fold risk for incident cardiovascular events (p=0.02) when all-cause death was excluded from the initially defined composite outcome.
Non-alcoholic steatohepatitis (NASH)
Wong et al. found that clonal hematopoiesis is associated with an increased risk of liver inflammation and chronic liver disease progression46. They attribute this to a dysregulated inflammatory response in the setting of CHIP. For this study, four large cohorts were combined, with orthogonal confirmation in a mouse model. The authors found evidence supporting a causal relationship between CHIP and the pathogenesis of liver disease, with a nearly two-fold increased risk of incidence of chronic liver disease if CHIP is found.
Chronic obstructive pulmonary disease (COPD)
A large study by Miller et al. investigated the relationship between CHIP and COPD, aggregating data from several cohorts of patients and a mouse model47. In their large primary cohort (COPDGene), 5.7% of patients showed CHIP mutations, the most common of which were DNMT3A, TET2 and ASXL1, with CHIP mutations more commonly observed in those with severe or very severe disease (OR 1.6).
Infections
Bolton et al. investigated the association between the presence of CHIP-related mutations, a pro-inflammatory immunologic profile, and worse outcomes for COVID-19, Clostridium difficile, Streptococcus and Enterococcus infections. In a cancer patient cohort, the presence of CH mutations was associated with severe infections by both C. difficile and Streptococcus/Enterococcus48. Increased secretion of interferon-gamma in chronic infection also appears to drive clonal hematopoiesis associated with DNMT3A loss of function49.
CHIP and COVID-19
The correlation between CHIP and COVID-19 is debated in the literature. Adopting mortality within 28 days of hospitalization for COVID-19 infection as an end-point, Miller et al. found no association between CHIP and COVID-1950. For this study, they focused on large CHIP clones with a VAF of 0.1 or greater, with the most commonly mutated genes being DNMT3A, TET2 and ASXL1, on a cohort of 1338 individuals drawn from two institutions. Conversely, Bolton et al. found that clonal hematopoiesis is associated with severe COVID-19 outcomes (p=0.01) in two separate cohorts of cancer and non-cancer patients from multiple institutions, totaling 525 individuals48. However, when stratifying the genes in the NGS panel the latter group used as putative drivers of CH and non-putative drivers, the association was not statistically significant (p 0.62). In contrast to the Miller et al. study which used overall survival, for both cohorts in the Bolton et al. study the outcome of severe COVID-19 was defined as hypoxia requiring more than 1 L of supplemental oxygen with documented hypoxia at an oxygen saturation of <94%, and the cohort of patients with cancer was adjusted for cancer primary site and exposure to cancer therapy. Overall, the authors found that the presence of CH, not specific alleles, is predictive of COVID-19 disease severity. Another interesting difference between the Bolton et al. and Miller et al. studies was that in the larger cohort of the Bolton study, the association of pre-existing CHIP (detected in blood specimens taken prior to July 1, 2019) was evaluated, rather than testing close to the time of COVID-19 diagnosis. One possibility is that an underlying increased inflammatory state (or tendency) in a subset of patients may lead to both greater likelihood of developing CHIP and greater likelihood of developing severe COVID-19. As above, Bolton et al. found that patients with CHIP also appear to be at risk for other infectious complications, including from Clostridium difficile and Streptococcus/Enterococcus. While not necessarily associated with increased risk of death, CHIP and infectious/inflammatory processes may further feed off of each other in a vicious cycle once developed to create more severe inflammatory symptoms, similar to as seen in atherosclerosis (Figure 1). Further studies will be needed to clarify this, and whether CHIP can be used as a potential biomarker for risk of severe infection/infectious complications in certain patient populations.
Is there any way to fight the expansion of or adverse outcomes from CHIP clones?
Administration of physiologic or supraphysiologic doses of vitamin C in mouse models of TET2-deficient hematopoiesis can activate residual TET2 function to reduce the expansion of mutated, clonal hematopoietic stem and progenitor cells51,52. While intriguing, preliminary studies on supraphysiological doses of vitamin C in humans have not found significant changes in the TET2 mutational allele burdens, although the cohorts were limited in number53. In a phase 2 clinical trial, Xie et al. found that there was an epigenetic impact on critical enhancer regions in hematopoietic stem cells in patients with stable disease, warranting further investigation53.
The reduction of IL-6 levels aiming at controlling the inflammatory burden is also being actively investigated in the literature. Bick et al. found that the presence of interleukin-6 receptor p.Asp358Ala alleles was associated with a reduction in the risk of cardiovascular events in patients with large CHIP clones, but not in those without. This allele is a genetic proxy for IL-6 pathway inhibition, leading the authors to pose that therapeutic inhibition of this pathway might be beneficial in patients with large CHIP clones to reduce cardiovascular risk45. The relationship between CHIP and IL-6 signaling was also explored in the randomized clinical trial CANTOS by Svensson et al54. The authors assessed the effect of administering Canakinumab, an anti-IL1β antibody, in patients with CHIP. They found that patients with CHIP in general had increased levels of IL-6, and that the blocking of IL1β on TET2-mutated CHIP cases had a statistically significant relative risk reduction on the occurrence of major adverse cardiovascular events on stratified analysis (p = 0.04) but not on interaction analysis (p = 0.14).
Can CHIP mutations ever be helpful in an inflammatory response? Emerging data on immune checkpoint blockade therapy and hematopoietic stem cell transplant
Recently, CHIP has been shown to influence T cell response in chronic states of antigenic stimulation, such as chronic infection and tumors. Kang et al. explored the effects of knock out DNMT3A, ASLX1 and TET2 on mouse models and found that these three genes control the developmental progression of multipotent progenitor T cells (Tpex) into terminally differentiated T cells (Tex), which are unresponsive to immune checkpoint blockade. Specifically, ASXL1 knock out cells were resistant to tumor microenvironment-induced exhaustion55.
Another emerging scenario involving CHIP is the consequences resulting from the engraftment of CH clones in recipients of hematopoietic stem cell transplantations (HSCT). Gibson et al. found that CH was present in 22.5% cases on a series of 1,727 donors56. Of these, 85% of donor clones showed engraftment in recipients, and CH involving DNMT3A at a VAF≥0.01 was associated with reduced relapse and increased chronic graft-versus-host-disease (GVHD). Frick et al. suggested that CH might enhance a generalized inflammatory response, contributing to both increased GVHD and graft-versus-leukemia responses. They also correlated CHIP with increased inflammation in recipients, proposing that the engrafted clonal monocytes (TET2 mutated) might promote a pro-inflammatory microenvironment, resulting in uncontrolled inflammation57. Overall, this remains an active area of investigation and clinical relevance due to the increasing age of HSCT donors and recipients, and increased life expectancy after HSCT, with arguments both for58 and against59 the current utility of screening for CH in hematopoietic stem cell donors.
Conclusion
The interplay of CHIP and various inflammatory conditions is multifaceted. There seems to be a potentially bidirectional relationship between CHIP and inflammation, involving many of the associated genes in both conditions, particularly in chronic inflammatory settings such as atherosclerotic disease. Additionally, some direct causal relationship instances have been suggested by the literature, including smoking, cancer therapy and chronic liver disease and potentially ischemic-origin CHF, although without definite association yet emerging with a particular type of inflammatory response.
In this context of what we know so far, at least a subset of patient populations may benefit from screening for CHIP and risk reduction strategies, including the possibility of targeted therapies60. While CHIP mutations are typically evaluated on peripheral blood specimens, abnormal inflammatory cells produced from the mutant clone can be detected in surgical pathology specimens, including cutaneous or other tissue biopsies of inflammatory lesions in VEXAS syndrome61 and atherosclerotic plaques in cardiovascular disease32. This broadens the potential for evaluating for CHIP in association with the inflammatory conditions in diagnostic pathology, which may have prognostic or treatment implications.
In particular, targeting candidate driver mutations in CHIP could present a therapeutic strategy that addresses multiple downstream inflammatory conditions, with compounding benefits. Overall, the association between CHIP, aging and inflammation is a rapidly evolving area with the potential for improved understanding and management to substantially decrease the morbidity associated with inflammatory and cardiovascular diseases.
References
- Steensma DP, Bejar R, Jaiswal S, et al. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood. 2015;126(1):9-16. doi:10.1182/blood-2015-03-631747
- Young AL, Challen GA, Birmann BM, Druley TE. Clonal haematopoiesis harbouring AML-associated mutations is ubiquitous in healthy adults. Nat Commun. 2016;7(1):12484. doi:10.1038/ncomms12484
- Jaiswal S, Libby P. Clonal haematopoiesis: connecting ageing and inflammation in cardiovascular disease. Nat Rev Cardiol. 2020;17(3):137-144. doi:10.1038/s41569-019-0247-5
- Challen GA, Sun D, Jeong M, et al. Dnmt3a is essential for hematopoietic stem cell differentiation. Nat Genet. 2012;44(1):23-31. doi:10.1038/ng.1009
- Jeong M, Park HJ, Celik H, et al. Loss of Dnmt3a Immortalizes Hematopoietic Stem Cells In Vivo. Cell Reports. 2018;23(1):1-10. doi:10.1016/j.celrep.2018.03.025
- Ley TJ, Ding L, Walter MJ, et al. DNMT3A Mutations in Acute Myeloid Leukemia. N Engl J Med. 2010;363(25):2424-2433. doi:10.1056/NEJMoa1005143
- Fujino T, Kitamura T. ASXL1 mutation in clonal hematopoiesis. Experimental Hematology. 2020;83:74-84. doi:10.1016/j.exphem.2020.01.002
- Jawad M, Afkhami M, Ding Y, et al. DNMT3A R882 Mutations Confer Unique Clinicopathologic Features in MDS Including a High Risk of AML Transformation. Front Oncol. 2022;12:849376. doi:10.3389/fonc.2022.849376
- Greenblatt SM, Nimer SD. Chromatin modifiers and the promise of epigenetic therapy in acute leukemia. Leukemia. 2014;28(7):1396-1406. doi:10.1038/leu.2014.94
- Chen JY, Luo CW, Lai YS, Wu CC, Hung WC. Lysine demethylase KDM2A inhibits TET2 to promote DNA methylation and silencing of tumor suppressor genes in breast cancer. Oncogenesis. 2017;6(8):e369. doi:10.1038/oncsis.2017.71
- Scourzic L, Mouly E, Bernard OA. TET proteins and the control of cytosine demethylation in cancer. Genome Med. 2015;7(1):9. doi:10.1186/s13073-015-0134-6
- Deplus R, Delatte B, Schwinn MK, et al. TET2 and TET3 regulate GlcNAcylation and H3K4 methylation through OGT and SET1/COMPASS. EMBO J. 2013;32(5):645-655. doi:10.1038/emboj.2012.357
- Ichiyama K, Chen T, Wang X, et al. The methylcytosine dioxygenase Tet2 promotes DNA demethylation and activation of cytokine gene expression in T cells. Immunity. 2015;42(4):613-626. doi:10.1016/j.immuni.2015.03.005
- Cull AH, Snetsinger B, Buckstein R, Wells RA, Rauh MJ. Tet2 restrains inflammatory gene expression in macrophages. Experimental Hematology. 2017;55:56-70.e13. doi:10.1016/j.exphem.2017.08.001
- Cai Z, Kotzin JJ, Ramdas B, et al. Inhibition of Inflammatory Signaling in Tet2 Mutant Preleukemic Cells Mitigates Stress-Induced Abnormalities and Clonal Hematopoiesis. Cell Stem Cell. 2018;23(6):833-849.e5. doi:10.1016/j.stem.2018.10.013
- Abdel-Wahab O, Adli M, LaFave LM, et al. ASXL1 mutations promote myeloid transformation through loss of PRC2-mediated gene repression. Cancer Cell. 2012;22(2):180-193. doi:10.1016/j.ccr.2012.06.032
- Kim N, Byun S, Um SJ. Additional Sex Combs-like Family Associated with Epigenetic Regulation. Int J Mol Sci. 2024;25(10):5119. doi:10.3390/ijms25105119
- Asada S, Fujino T, Goyama S, Kitamura T. The role of ASXL1 in hematopoiesis and myeloid malignancies. Cell Mol Life Sci. 2019;76(13):2511-2523. doi:10.1007/s00018-019-03084-7
- Yang H, Kurtenbach S, Guo Y, et al. Gain of function of ASXL1 truncating protein in the pathogenesis of myeloid malignancies. Blood. 2018;131(3):328-341. doi:10.1182/blood-2017-06-789669
- Nakad R, Schumacher B. DNA Damage Response and Immune Defense: Links and Mechanisms. Front Genet. 2016;7. doi:10.3389/fgene.2016.00147
- Zhang CRC, Nix D, Gregory M, et al. Inflammatory cytokines promote clonal hematopoiesis with specific mutations in ulcerative colitis patients. Experimental Hematology. 2019;80:36-41.e3. doi:10.1016/j.exphem.2019.11.008
- Chen J, Nie D, Wang X, et al. Enriched clonal hematopoiesis in seniors with dietary vitamin C inadequacy. Clin Nutr ESPEN. 2021;46:179-184. doi:10.1016/j.clnesp.2021.10.014
- Haring B, Reiner AP, Liu J, et al. Healthy Lifestyle and Clonal Hematopoiesis of Indeterminate Potential: Results From the Women’s Health Initiative. JAHA. 2021;10(5):e018789. doi:10.1161/JAHA.120.018789
- Pencina MJ, D’Agostino RB, Larson MG, Massaro JM, Vasan RS. Predicting the 30-Year Risk of Cardiovascular Disease: The Framingham Heart Study. Circulation. 2009;119(24):3078-3084. doi:10.1161/CIRCULATIONAHA.108.816694
- Welch JS, Ley TJ, Link DC, et al. The Origin and Evolution of Mutations in Acute Myeloid Leukemia. Cell. 2012;150(2):264-278. doi:10.1016/j.cell.2012.06.023
- Bolton KL, Ptashkin RN, Gao T, et al. Cancer therapy shapes the fitness landscape of clonal hematopoiesis. Nat Genet. 2020;52(11):1219-1226. doi:10.1038/s41588-020-00710-0
- Ferrucci L, Corsi A, Lauretani F, et al. The origins of age-related proinflammatory state. Blood. 2005;105(6):2294-2299. doi:10.1182/blood-2004-07-2599
- Perdigones N, Perin JC, Schiano I, et al. Clonal hematopoiesis in patients with dyskeratosis congenita. American J Hematol. 2016;91(12):1227-1233. doi:10.1002/ajh.24552
- Hinds DA, Barnholt KE, Mesa RA, et al. Germ line variants predispose to both JAK2 V617F clonal hematopoiesis and myeloproliferative neoplasms. Blood. 2016;128(8):1121-1128. doi:10.1182/blood-2015-06-652941
- Schmidt V, Hogan AE, Fallon PG, Schwartz C. Obesity-Mediated Immune Modulation: One Step Forward, (Th)2 Steps Back. Front Immunol. 2022;13:932893.doi:10.3389/fimmu.2022.932893
- Donath MY, Shoelson SE. Type 2 diabetes as an inflammatory disease. Nat Rev Immunol. 2011;11(2):98-107. doi:10.1038/nri2925
- Jaiswal S, Fontanillas P, Flannick J, et al. Age-Related Clonal Hematopoiesis Associated with Adverse Outcomes. N Engl J Med. 2014;371(26):2488-2498. doi:10.1056/NEJMoa1408617
- Jaiswal S, Natarajan P, Silver AJ, et al. Clonal Hematopoiesis and Risk of Atherosclerotic Cardiovascular Disease. N Engl J Med. 2017;377(2):111-121. doi:10.1056/NEJMoa1701719
- Heyde A, Rohde D, McAlpine CS, et al. Increased stem cell proliferation in atherosclerosis accelerates clonal hematopoiesis. Cell. 2021;184(5):1348-1361.e22. doi:10.1016/j.cell.2021.01.049
- Lee J, Taneja V, Vassallo R. Cigarette smoking and inflammation: cellular and molecular mechanisms. J Dent Res. 2012;91(2):142-149. doi:10.1177/0022034511421200
- Elisia I, Lam V, Cho B, et al. The effect of smoking on chronic inflammation, immune function and blood cell composition. Sci Rep. 2020;10(1):19480. doi:10.1038/s41598-020-76556-7
- Savola P, Lundgren S, Keränen MAI, et al. Clonal hematopoiesis in patients with rheumatoid arthritis. Blood Cancer Journal. 2018;8(8):69. doi:10.1038/s41408-018-0107-2
- Beck DB, Ferrada MA, Sikora KA, et al. Somatic Mutations in UBA1 and Severe Adult-Onset Autoinflammatory Disease. N Engl J Med. 2020;383(27):2628-2638. doi:10.1056/NEJMoa2026834
- Gutierrez-Rodrigues F, Kusne Y, Fernandez J, et al. Spectrum of clonal hematopoiesis in VEXAS syndrome. Blood Journal. Published online April 21, 2023:blood.2022018774. doi:10.1182/blood.2022018774
- Gurnari C, Pascale MR, Vitale A, et al. Diagnostic capabilities, clinical features, and longitudinal UBA1 clonal dynamics of a nationwide VEXAS cohort. American J Hematol. 2024;99(2):254-262. doi:10.1002/ajh.27169
- Bolton KL, Ptashkin RN, Gao T, et al. Cancer therapy shapes the fitness landscape of clonal hematopoiesis. Nat Genet. 2020;52(11):1219-1226. doi:10.1038/s41588-020-00710-0
- Yvan-Charvet L, Pagler T, Gautier EL, et al. ATP-binding cassette transporters and HDL suppress hematopoietic stem cell proliferation. Science. 2010;328(5986):1689-1693. doi:10.1126/science.1189731
- Murphy AJ, Tall AR. Disordered haematopoiesis and athero-thrombosis. Eur Heart J. 2016;37(14):1113-1121. doi:10.1093/eurheartj/ehv718
- Dorsheimer L, Assmus B, Rasper T, et al. Association of Mutations Contributing to Clonal Hematopoiesis With Prognosis in Chronic Ischemic Heart Failure. JAMA Cardiol. 2019;4(1):25. doi:10.1001/jamacardio.2018.3965
- Bick AG, Pirruccello JP, Griffin GK, et al. Genetic Interleukin 6 Signaling Deficiency Attenuates Cardiovascular Risk in Clonal Hematopoiesis. Circulation. 2020;141(2):124-131. doi:10.1161/CIRCULATIONAHA.119.044362
- Wong WJ, Emdin C, Bick AG, et al. Clonal haematopoiesis and risk of chronic liver disease. Nature. 2023;616(7958):747-754. doi:10.1038/s41586-023-05857-4
- Miller PG, Qiao D, Rojas-Quintero J, et al. Association of clonal hematopoiesis with chronic obstructive pulmonary disease. Blood. 2022;139(3):357-368. doi:10.1182/blood.2021013531
- Bolton KL, Koh Y, Foote MB, et al. Clonal hematopoiesis is associated with risk of severe Covid-19. Nat Commun. 2021;12(1):5975. doi:10.1038/s41467-021-26138-6
- Hormaechea-Agulla D, Matatall KA, Le DT, et al. Chronic infection drives Dnmt3a-loss-of-function clonal hematopoiesis via IFNγ signaling. Cell Stem Cell. 2021;28(8):1428-1442.e6. doi:10.1016/j.stem.2021.03.002
- Miller PG, Fell GG, Foy BH, et al. Clonal hematopoiesis of indeterminate potential and risk of death from COVID-19. Blood. 2022;140(18):1993-1997. doi:10.1182/blood.2022018052
- Agathocleous M, Meacham CE, Burgess RJ, et al. Ascorbate regulates haematopoietic stem cell function and leukaemogenesis. Nature. 2017;549(7673):476-481. doi:10.1038/nature23876
- Cimmino L, Dolgalev I, Wang Y, et al. Restoration of TET2 Function Blocks Aberrant Self-Renewal and Leukemia Progression. Cell. 2017;170(6):1079-1095.e20. doi:10.1016/j.cell.2017.07.032
- Xie Z, Fernandez J, Lasho T, et al. High-dose IV ascorbic acid therapy for patients with CCUS with TET2 mutations. Blood. 2024;144(23):2456-2461. doi:10.1182/blood.2024024962
- Svensson EC, Madar A, Campbell CD, et al. TET2-Driven Clonal Hematopoiesis and Response to Canakinumab: An Exploratory Analysis of the CANTOS Randomized Clinical Trial. JAMA Cardiol. 2022;7(5):521-528. doi:10.1001/jamacardio.2022.0386
- Kang TG, Lan X, Mi T, et al. Epigenetic regulators of clonal hematopoiesis control CD8 T cell stemness during immunotherapy. Science. 2024;386(6718):eadl4492. doi:10.1126/science.adl4492
- Gibson CJ, Kim HT, Zhao L, et al. Donor Clonal Hematopoiesis and Recipient Outcomes After Transplantation. J Clin Oncol. 2022;40(2):189-201. doi:10.1200/JCO.21.02286
- Frick M, Chan W, Arends CM, et al. Role of Donor Clonal Hematopoiesis in Allogeneic Hematopoietic Stem-Cell Transplantation. J Clin Oncol. 2019;37(5):375-385. doi:10.1200/JCO.2018.79.2184
- DeZern AE, Gondek LP. Stem cell donors should be screened for CHIP. Blood Adv. 2020;4(4):784-788. doi:10.1182/bloodadvances.2019000394
- Gibson CJ, Lindsley RC. Stem cell donors should not be screened for clonal hematopoiesis. Blood Adv. 2020;4(4):789-792. doi:10.1182/bloodadvances.2019000395
- Marnell CS, Bick A, Natarajan P. Clonal hematopoiesis of indeterminate potential (CHIP): Linking somatic mutations, hematopoiesis, chronic inflammation and cardiovascular disease. Journal of Molecular and Cellular Cardiology. 2021;161:98-105. doi:10.1016/j.yjmcc.2021.07.004
- Zakine E, Schell B, Battistella M, et al. UBA1 Variations in Neutrophilic Dermatosis Skin Lesions of Patients With VEXAS Syndrome. JAMA Dermatol. 2021;157(11):1349-1354. doi:10.1001/jamadermatol.2021.3344

Marcello Pecoraro Toscano, MD, is currently a third-year resident in Anatomic and Clinical Pathology at Cleveland Clinic, Cleveland, OH. He is a junior member on the College of American Pathologists Personalized Healthcare Committee.

Genevieve M. Crane, MD, PhD, FCAP is an associate professor of pathology at Cleveland Clinic Lerner College of Medicine at Case Western Reserve University in Cleveland, OH. Her clinical roles include staff hematopathologist, medical director of manual hematology and associate medical director of the Immunohistochemistry Laboratory at the Cleveland Clinic. She received a B.S. summa cum laude in Chemical Engineering from Rice University, an M.Phil. from University College London as a British Marshall Scholar, and M.D. and Ph.D. degrees from the University of Michigan through the Medical Scientist Training Program. She did postdoctoral work at MIT in cancer biology before completing a residency in Anatomic Pathology and a fellowship in Hematopathology at Johns Hopkins Hospital. Dr. Crane has authored more than 70 journal articles and has 2 related patents. She is author of numerous book chapters, two textbooks (Survival Guide to Lymph Node Pathology [Innovative Science Press, 2021] and Biopsy Interpretation of the Bone Marrow [Wolters Kluwer, 2023]), and co-editor of Precision Molecular Pathology of Aggressive B-cell Lymphomas (Springer, 2023). She is section editor for Hematopathology at Archives of Pathology and Laboratory Medicine, deputy-editor-in-chief covering the hematopathology topics at PathologyOutlines.com, the world’s largest online pathology resource, serves on multiple other editorial boards, and is actively involved in multiple pathology societies. She is passionate about developing novel methods for pathology education and outreach and improving diagnostic pathology and disease understanding through research. In her spare time, she enjoys figure skating, spending time with her son, and being outdoors.