- Home
- Member Resources
- Articles
- Mismatches in Mismatch Repair: Navigating Discordant MMR/MSI Test Results
Introduction
Testing for mismatch repair (MMR) deficiency and/or microsatellite instability (MSI) involves immunohistochemistry (IHC), polymerase chain reaction (PCR)-based assays, and next-generation sequencing (NGS). Currently, the three main testing approaches are IHC for the MMR proteins, PCR-based MSI assays, and NGS panels that can determine both MSI status and pathologic mutations in the MMR genes. This article compares these methods and offers guidance for resolving conflicting results.
Background
The DNA MMR system maintains genomic fidelity by correcting base substitution mismatches and insertion-deletion loops that arise during DNA replication and recombination. This process is primarily mediated by a heterodimeric protein complex made of the key proteins MLH1, MSH2, MSH6, and PMS2. Germline or somatic inactivation of the genes encoding these proteins leads to deficient MMR (dMMR), resulting in the accumulation of somatic mutations at a rate several orders of magnitude higher than in MMR-proficient cells. The molecular hallmark of this hypermutable phenotype is MSI, characterized by length variations in short tandem DNA repeats. Clinically, dMMR/MSI status has emerged as a critical marker of potential response and eligibility for immune checkpoint inhibitor (ICI) therapy.
Methods for MMR/MSI Detection
MMR protein detection via IHC is a commonly used, fast, and inexpensive method for assessing MMR status in FFPE tumor tissue. It works by detecting four key MMR proteins: MLH1, PMS2, MSH2, and MSH6, which function as obligate heterodimers. MLH1 pairs with PMS2, and MSH2 pairs with MSH6. Loss of MLH1 leads to loss of PMS2, and loss of MSH2 leads to loss of MSH6; however, the reverse is not true. Interpretation involves determining whether nuclear expression of these four proteins is completely lost in tumor cells, as assessed by IHC, relative to adjacent nontumor cells, which serve as the internal positive control. A tumor is classified as MMR-proficient (pMMR) if all four proteins show intact nuclear staining in the tumor cells. Conversely, a tumor is classified as dMMR if there is a complete and unequivocal loss of nuclear staining for one or more MMR proteins in tumor cells. Using MMR IHC has several advantages, including a short turnaround time (typically one to two days), relative affordability, and broad availability in most laboratories.
Additionally, MMR IHC identifies the likely mutated MMR gene, enabling targeted germline testing. Its main drawback is that it assesses protein presence rather than function, which can lead to a false-negative report of pMMR, especially when there is a missense mutation in the C-terminus of the MMR gene. Other issues include loss of MMR protein expression resulting from poor fixation or epitope-escaping variants, as well as heterogeneous staining. Finally, there is a lack of standardized protocols between laboratories. Nevertheless, MMR IHC remains a workhorse for MMR/MSI screening, given its speed and informative output.1
MSI testing assays evaluate for the functional consequences of MMR deficiency. Some of the PCR-based tests do this by comparing the mutational status of five or more small microsatellite repeats in the patient’s tumor DNA with that in their normal tissue. In contrast, some newer PCR-based assays and nearly all NGS-based assays use only tumor tissue, typically employing up to 13 microsatellite markers with PCR-based methods and hundreds to thousands of microsatellite markers with NGS-based assays. Limitations of PCR-based MSI testing include the requirement for matched normal tissue, longer turnaround times, and varying sensitivity across different tumor types. While PCR-based MSI testing is mostly concordant with MMR IHC in colorectal cancer, lower concordance has been observed in endometrial cancer.2,3 The concordance with MMR IHC in less frequently tested tissue types, such as the upper GI tract, prostate, and ovary, remains unclear. The four most common PCR-based MSI testing assays are compared in Table 1.
Table 1. PCR-based MSI assays at a glance: legacy and contemporary options with key features.
| Assay Feature | Bethesda Panel (Traditional) | Promega MSI Analysis System | Idylla™ MSI Test (Biocartis) | TrueMark™ MSI Assay (Thermo Fisher) |
|---|---|---|---|---|
| Primary Use | Historical Standard | Current Gold Standard | Rapid, Automated Testing | High-Throughput Research/Clinical |
| Marker Type | Mononucleotide & Dinucleotide | Mononucleotide (plus 2 Pentanucleotides for QC) | Mononucleotide | Mononucleotide |
| Number of Markers | 5 | 5 (for MSI) + 2 (for QC) | 7 | 13 |
| Matched Normal Required? | Yes | Yes | No | No |
| Technology | PCR + Capillary Electrophoresis | PCR + Capillary Electrophoresis | Fully Automated Real-Time PCR | PCR + Capillary Electrophoresis |
| Workflow | More complex, manual | More complex, manual | Simple, fast, automated | Simplified, high-throughput |
NGS-based assays can also be used to test for MSI. Common NGS-based MSI algorithms are MSISensor and MANTIS, which are designed to detect instability across multiple loci. Moreover, NGS assays can be efficiently designed to sequence the MMR genes, enabling simultaneous identification of pathogenic variants in one or more MMR genes. NGS assays can also simultaneously assess for pathologic variants in BRAF V600E and POLE/POLD1. Finally, NGS assays can provide the tumor mutational burden (TMB) of a specimen. NGS-based MSI testing provides a comprehensive genomic view from a single test and is particularly efficient when patients require simultaneous large-panel NGS-based DNA assays for tumor profiling.4 The performance of MSI testing with NGS-based assays depends on the panel design and validation; however, modern datasets demonstrate noninferiority to IHC and strong concordance across various tumor types.5,6 Real-world and pathological series also document rare cases that each modality can miss, reinforcing the need for orthogonal testing in equivocal scenarios.5,7 Each method for MMR/MSI testing is compared in Table 2.
Table 2. Comparison of methods for MMR/MSI testing.
| IHC (MMR Protein) | PCR-Based MSI | NGS-Based MSI | |
|---|---|---|---|
| What it detects | Presence or absence of MMR proteins (MLH1, MSH2, MSH6, PMS2) in tumor cell nuclei (expression status) | Compares length changes in specific microsatellite loci in tumor vs normal DNA (functional readout) | Compares length changes across many (hundreds to thousands) microsatellite loci; can also detect mutations in MMR genes (if performed with a gene panel) |
| Principle | Antibody staining on FFPE tissue; complete loss of nuclear staining in tumor cells with intact internal controls indicates dMMR. | PCR amplification of microsatellite repeats (typically 5–13 markers). Instability is the presence of new allele sizes in tumor DNA that are not present in normal DNA. | Targeted, exome, or whole genome sequencing with a computational algorithm that quantifies instability at numerous loci. |
| Sample type | FFPE slides | Tumor DNA (FFPE acceptable), usually plus matched normal control (whole blood or normal tissue). Newer assays do not require a normal matched control. | Tumor DNA (FFPE acceptable). Matched normal is often not required for MSI calling |
| Turnaround time | Fast: ≈1–3 days | Moderate: several days to ≈2 weeks | Longer: ≈1–3 weeks, including analysis |
| Cost | Low | Moderate | Higher (mitigated when bundled with broader panel testing) |
| Information provided | Identifies which MMR protein(s) are lost, guiding methylation/germline reflex testing | Confirms functional consequence of MMR protein loss/mutation (MSI status) | Comprehensive genomic profile: MSI status, identification of MMR gene variants, along with BRAF V600E status (CRC), POLE/POLD1 status, and tumor mutation burden (TMB) |
| Advantages | Widely available, quick, inexpensive; can be done on a tiny tumor sample; no matched normal tissue required; pinpoints the likely gene defect for Lynch workup | Longstanding clinical validation; high specificity for MSI-H in CRC; direct phenotype readout | One test answers many questions, demonstrating strong concordance with IHC/PCR in validated panels, and can detect cases missed by limited-marker PCR or IHC5,7 |
| Limitations | Indirect functional readout; rare false negatives (nonfunctional expressed protein); artifacts/heterogeneity can complicate calls.26 Rare cases of Constitutional Mismatch Repair Deficiency (CMMRD) syndrome, where the patient’s normal cells (internal control) will lack staining for one MMR protein | Requires matched normal tissue in most commonly used assay; requires at least 20% tumor content in provided sample; limited markers; variable sensitivity outside of CRC1 | Typically requires at least 20% tumor content in the provided sample; requires resources/bioinformatics; performance varies by panel and tumor type; longer TAT4 |
Discordance of MSI Testing Results
Since the first reports of MSI in 1993 and the launch of the Bethesda panel in 1997,8–11 MMR/MSI testing has evolved from a niche hereditary cancer screen to a pan-cancer companion diagnostic that guides chemotherapy choices and, critically, eligibility for ICIs. Early comparisons of IHC and PCR in colorectal cohorts showed low discordance (approximately 1% to 3%), but these studies relied on suboptimal methods, notably 2-antibody IHC (MLH1/MSH2) and dinucleotide-heavy PCR panels; so microsatellite instability-high (MSI-H) tumors with isolated PMS2 or MSH6 loss were often mislabeled as pMMR.12,13 Transitioning to 4-antibody IHC and mononucleotide-rich PCR improved sensitivity; Hatch et al. highlighted this shift and the need for mononucleotide markers.14,15
Contemporary data reveal that "discordance" depends on technique, interpretation, and tumor biology. Real-world rates range from well below 1% in tightly controlled series to double-digit levels when initial reads and community practice are considered; one study found 10% misclassification among locally called dMMR/MSI-H metastatic colorectal cancer (mCRC) cases and linked these errors to primary ICI nonresponse.16 Other series note modest cross-platform assay discordance,17 while an upper-bound report of 19.3% underscores how workflow and context can inflate discrepancies.18 Rates may be higher outside CRC (e.g., endometrial cancer), where patterns like isolated MSH6 loss are more common.19 Overall, discordance is not a biological constant, but rather a function of assay design, pathologist review rigor, and cohort heterogeneity, which suggests the need for orthogonal testing and expert adjudication when results do not align.
Current MSI/MMR Assays and Causes of Discordance
Currently, the causes of discordant results can be systematically categorized into four major groups: (1) technical and interpretive artifacts; (2) differences in phenotypic (IHC) and genotypic (PCR/NGS) assays; (3) multiple primary tumors; and (4) idiopathic causes. These scenarios are ordered from the most common to the rarest causes of discordance. The first three scenarios represent resolvable discordance. In contrast, the fourth scenario, although likely rare, is an area that has not received significant attention and currently lacks a straightforward, protocol-driven approach.
Technical and interpretive artifacts include:
- Suboptimal fixation: This can lead to weak or absent MMR IHC staining, resulting in an erroneous interpretation of protein loss. In larger tissue samples, a specific area with poor formalin fixation may occur (typically in the center of the tissue), mimicking focal clonal loss of MMR proteins. Typically, this area does not stain with any of the MMR proteins, and if any normal tissue is present, the normal tissue (internal control) will also show loss of MMR IHC staining.
- Low tumor cellularity: This can cause false-negative microsatellite stable (MSS) results in PCR/NGS by masking the unstable microsatellite alleles.20
- Prior chemo/radiation therapy: This can induce MMR protein expression loss, particularly MSH6, creating a dMMR/MSS phenotype that may not reflect the patient’s tumor baseline status.21,22 When feasible, assess pretreatment tissue. If only post-treatment tissue is available and the results are unexpected, confirm the results with an orthogonal method or an alternative block.23
- Interpretive error: Subjective IHC interpretation and misinterpretation of MSI-PCR electropherograms contribute to interpretive discordance. Consensus conferences or experienced colleagues can provide valuable assistance. Misinterpreting "intact" IHC staining, when there is heterogeneous loss, can lead to false negatives.
- IHC antibody clone-specific issues: Rare germline polymorphisms can alter the epitope recognized by a specific antibody clone, leading to false protein loss. A known example is the MLH1 p.V384D variant, which is missed by the MLH1 G168-15 antibody clone.23
Less Common Patterns of MMR IHC Loss
Heterogeneous Loss of Expression of One or More of the MMR IHC Proteins
In contrast to the binary classification of MMR protein expression as either uniformly intact or completely absent, a subset of tumors exhibits heterogeneous staining for one or more MMR proteins. Heterogeneous MMR protein expression can manifest in several distinct patterns, specifically:
- Subclonal: geographically distinct region(s) of intact and lost staining
- Focal: small clusters of cells with loss
- Zonal: distinct areas or patches within the tumor where the MMR protein is absent
- Mosaic: intermixed single cells with variable staining
Some laboratories assess only MMR protein expression by IHC without concurrent MSI testing by PCR or NGS. A large retrospective study of more than 4,400 patients found that approximately 50% of cases with heterogeneous IHC loss were MSI-H by PCR, whereas the remaining cases were MSS or, less frequently, microsatellite instability-low (MSI-L).24 Patients with heterogeneous MMR protein loss, therefore, have approximately a 50% chance of being MSI-H and eligible for ICI therapy. This means that all patients with heterogeneous MMR protein loss on IHC require microsatellite testing (PCR or NGS), specifically sampling the area with MMR IHC loss.2,25 Heterogeneous loss also nearly doubles the risk of the patient having Lynch syndrome, necessitating appropriate germline testing and follow-up.19
Heterogeneous MMR IHC With Synchronous MSI-H Results
Microdissection-based studies demonstrate that IHC-negative foci often harbor MSI-H and/or driver events, such as MLH1 methylation or an MSH6 frameshift mutation. At the same time, the adjacent IHC-intact areas are MSS, confirming biologic subclonality.26 These findings indicate that the heterogeneous patterns of MMR protein expression reflect genuine intratumoral molecular diversity rather than a staining artifact; therefore, reporting heterogeneous loss and the pattern explicitly (e.g., "subclonal MSH6 loss in approximately 10% of tumor cells") helps drive appropriate follow-up.
Heterogeneous MMR IHC With Synchronous MSI-L or MSS Results
Interpreting MMR/MSI testing in patients with heterogeneous MMR protein loss is challenging, particularly for determining eligibility for immune checkpoint therapy. If circling an area for MSI-testing, one must circle an area with MMR IHC loss. When reviewing MSI testing results, it is critical to determine whether the area with MMR IHC loss was tested. If only an intact MMR IHC area was tested for MSI, retesting the area with the greatest MMR IHC loss using PCR- or NGS-based MSI is recommended.
Suppose concordant or follow-up PCR-based MSI testing of the areas with MMR protein loss shows MSS or MSI-L. In that case, further NGS testing of the tumor (specifically the area of loss) for MSI status and pathogenic MMR gene mutations can be considered. If NGS testing classifies the tumor as MSI-H and/or identifies two pathogenic MMR variants (or one variant and with loss of heterozygosity [LOH] of that gene), this supports an MSI-H classification of the tumor. Conversely, if NGS still classifies the tumor as MSS and no pathogenic mutations in MMR genes are found, a final interpretation of MSS classification is supported; however, there is limited information on how these rare patients respond to immune checkpoint inhibition, highlighting the need for further studies.
Complete Loss of All Four MMR Proteins on IHC: The "Null" Phenotype
Although rare, the complete absence of all four MMR proteins is termed the null phenotype. This complex pattern can result from concurrent alterations in both the MLH1 and MSH2 pathways, such as a germline MSH2 mutation combined with somatic MLH1 promoter hypermethylation27; however, calling this phenotype dMMR based solely on IHC staining is not advised. Other studies should be done, including the use of different antibody clone(s), MLH1 promoter methylation assays, and/or an NGS-based panel to support an interpretation of a null phenotype.
Discordance Due to Differences in Phenotypic (IHC) and Genotypic (PCR/NGS) Assays
In these cases, the interpretation of each assay is correct; however, the discordance arises from whether the assay evaluates protein expression or genetic mutations at the DNA level.
pMMR by IHC/MSI-H by Molecular Testing:
- pMMR can arise when an MMR gene harbors a mutation that abrogates its DNA repair function but does not prevent the production of a full-length, antigenically intact protein. These are typically non-truncating C-terminal missense mutations, with the classic example seen in MSH6.25,26
- Pathogenic mutation in the exonuclease domain of DNA polymerase epsilon (POLE) or delta 1 (POLD1). These mutations create an ultramutated phenotype. The majority of POLE mutant tumors are MSS; however, some can have secondary MSI-H due to overwhelming the microsatellite repair system.28 In these cases, where the MMR proteins are intact and no pathogenic mutations are present, the IHC result will be pMMR. Additionally, pathogenic POLE-mutant cancers can lead to secondary mutations in the MMR genes, resulting in an MSI-H phenotype with dMMR; however, these cases may exhibit atypical loss patterns on MMR IHC, which can be a clue to a POLE mutation.
- Noncanonical Gene Involvement: The standard four-protein IHC panel only assesses the core MMR proteins; however, the MMR machinery involves other gene products, including MSH3, PMS1, MLH3, and EXO1.30–32 Pathogenic mutations in these genes may cause MMR dysfunction and an MSI-H phenotype, which PCR detects; however, IHC for the main four proteins is normal (pMMR).
dMMR by IHC/MSS by Molecular Testing:
- MSH6 Deficiency: The primary biological driver of this phenotype is often pathologic variants in MSH6, which lead to nonsense-mediated decay of MSH6 RNA and loss of MSH6 protein expression. Currently, it is thought that the loss of MSH6 gene expression leads to a less profound MSI phenotype, characterized by mutations primarily in mononucleotide repeats and smaller allelic shifts that may fall below the detection threshold of standard PCR panels, resulting in an MSS or MSI-L result.29 In discordant cases with isolated MSH6 loss but MSS or only minimal microsatellite shifts, which is particularly common in endometrial carcinoma and may also be seen in post-treatment specimens, NGS-based MSI detection may still identify MSI when IHC or PCR-MSI is falsely negative.25,29
- Please see the Tumor-Specific Considerations section (Endometrial Carcinoma) below for additional discussion and assay-performance considerations in this setting.
Multiple Primary Tumors
In this scenario, a primary tumor may be correctly identified as dMMR/MSI-H. Still, a subsequent biopsy of a metastatic lesion for MMR/MSI analysis may sample a metastasis from a second, synchronous, or metachronous primary tumor with pMMR/MSS. Geurts et al. found that multiple primary tumors accounted for 23% (3 of 13) of their initial discordant cases, none of whom benefited from ICI therapy, highlighting this as a critical diagnostic pitfall.33 As blood-based liquid biopsies are increasingly utilized, many of which test for MSI status, consideration of multiple primary tumors is necessary when there is discordance between MMR/MSI testing in the presumed primary tumor and the liquid biopsy results.
Idiopathic Discordance
Discordant cases that are dMMR or MSI-H but lack two pathogenic MMR gene variants warrant a systematic investigation. Initially, one must confirm the adequacy of internal controls for MMR IHC. Subsequently, consultation with a molecular pathologist is crucial to determine whether the NGS assay provides evidence for a "second hit," such as LOH or homozygous deletion in the MMR gene, corresponding to the protein loss observed on IHC.
However, discrepancies may persist because not all NGS panels are designed or validated to detect LOH, homozygous deletions, large structural variants, copy number changes, or deep intronic splice variants.34 Beyond these assay limitations, other biological mechanisms can potentially cause MMR deficiency.32 For example, upregulation of specific microRNAs can transcriptionally silence MMR genes,35 while pathogenic variants in related genes, such as EXO1, can disrupt the repair complex and induce an MSI-H phenotype.34 Finally, preanalytical factors, including prolonged tumor ischemia, delayed fixation, or hypoxia, can cause artifactual loss of MMR protein expression, mimicking a true dMMR result.36
Approach to Discordant Cases:
- Expert Review: All discordant or ambiguous results should undergo expert pathology review to rule out interpretive errors or technical artifacts, such as poor fixation.
- Consider the Clinical Context: Account for prior neoadjuvant therapy, which can lead to loss of MMR protein. Whenever possible, test pretreatment tissue.
- Report heterogeneous MMR IHC loss: Describe the pattern and consider microdissection-based MSI testing on the area with MMR protein loss.2,37
- Discuss at a Molecular Tumor Board: Complex cases benefit from multidisciplinary input to arrive at an integrated diagnosis.
- Final Clinical Interpretation: After excluding resolvable causes, any definitive evidence of MMR deficiency (either dMMR by IHC or MSI-H by PCR/NGS) should be considered a positive result for determining eligibility for ICI therapy.2
A suggested approach to interpreting discordant or unusual MMR/MSI patterns, along with possible next steps, is covered in Table 3.
Table 3. Interpreting discordant or unusual MMR/MSI result patterns and possible next steps:
| Tumor MMR/MSI Profile (IHC vs MSI status) | Potential Underlying Cause(s) | Lynch Syndrome Risk? | Possible Next Steps / Workup |
|---|---|---|---|
| pMMR by IHC; MSI-H | Expressed but nonfunctional MMR protein (most commonly MSH6); POLE ultramutator phenotype; noncanonical MMR gene mutation (see text) | Often elevated risk if not POLE mutated and negative for hypermethylation of MLH1 promoter. | MLH1 promoter methylation testing; POLE/POLD1 sequencing of tumor; MMR gene sequencing of tumor tissue (likely in a large NGS panel) with follow-up germline testing if an MMR gene is found to be mutated |
| dMMR by IHC with combined MLH1/PMS2 loss; MSS | MLH1 promoter hypermethylation; IHC artifact; insensitive MSI assay; early acquired dMMR mutation | Low if MLH1 promoter is hypermethylated; higher if MLH1 promoter is unmethylated; high if young/family history positive | Verify IHC; MLH1 methylation assay ± BRAF V600E testing (in CRC); alternate MSI assay; MMR gene sequencing of tumor tissue (likely in a large NGS panel) with germline testing if MLH1 or PMS2 mutation identified |
| dMMR by IHC with combined MSH2/MSH6 loss; MSS or MSI-L | MSH2 mutation with "weaker" MSI-H phenotype (germline or somatic); rarely can have germline or somatic MSH6 mutation; POLE hypermutator phenotype | Moderate–High | Strongly consider germline MSH2 and MSH6 testing; retest with a different MSI assay; test the tumor for POLE/POLD1 mutation; MMR gene sequencing of tumor tissue (likely in a large NGS panel)2,25 |
| dMMR by IHC with isolated MSH6 loss; MSS or MSI-Low | MSH6 mutation with a "weaker" MSI-H phenotype (germline more likely than somatic) | High, more so if young/positive history | Strongly consider germline MSH6 testing; MMR gene sequencing of tumor tissue (likely in a large NGS panel) |
| dMMR by IHC with isolated PMS2 loss; MSS or MSI-Low | PMS2 mutation (germline or somatic); rarely, it can result from a non-truncating MLH1 mutation that affects PMS2 binding.54,55 | Elevated without other viable explanation | Strongly consider germline PMS2 testing; confirm PMS2 loss on another block; MMR gene sequencing of tumor tissue (likely in a large NGS panel) |
| Heterogeneous/subclonal MLH1/PMS2 loss; MSS or MSI-H | Subclonal MLH1 promoter hypermethylation (usually MSS); subclonal MLH1 mutation (can be MSS or MSI-H) | Low if MLH1 promoter is hypermethylated; increased if unmethylated/younger | Microdissect area(s) with loss for MSI and/or MLH1 promoter methylation testing |
| Heterogeneous/subclonal MSH6 loss; MSS or MSI-H | Somatic MSH6 variants enriched in subclone; possible germline MSH6 mutation; previous chemoradiation therapy21,22 | Moderate | MSI testing on MSH6 IHC-negative foci; germline MSH6 testing as indicated by context26 |
| Non-canonical/unusual pattern of MMR protein loss (most common is MLH1, PMS2, and MSH6 loss); MSS (most common) or MSI-H (less common) | Two separate mutation events secondary to MSI-H phenotype leading to second hit (most commonly in MSH656) or a hypermutation phenotype (POLE/POLD1 pathogenic mutation); tumor heterogeneity; previous chemoradiation therapy (most commonly MSH6 loss21,22) | Often elevated risk if not POLE/POLD1 mutated and/or negative for hypermethylation of MLH1 promoter | Test tumor for POLE/POLD1 mutation; MMR gene sequencing of tumor tissue (likely in a large NGS panel); MLH1 promoter methylation testing |
Tumor-Specific Considerations:
- Colorectal Carcinoma: IHC, PCR, and NGS are well-validated and accepted methods that offer largely concordant results2; however, combining two or more methods will help to identify microsatellite repair-deficient tumors that can be missed using only one method alone.24,38
- Gastroesophageal and Small Bowel Carcinoma: Contrary to previous guidance,2 a recent large-scale study5 demonstrated that NGS-based MSI testing is noninferior to MMR IHC and PCR-based MSI testing in these tumor types.
- Endometrial Carcinoma: Testing endometrial carcinomas solely with PCR-based MSI testing is no longer recommended,2 as multiple studies have demonstrated that it lacks sensitivity in these types of tumors.6,38,39 Previous guidelines2 also advised against using NGS-based MSI testing alone; however, a 2024 study showed that a novel NGS-based MSI detection method (MSIPeak) exhibited higher sensitivity than PCR for detecting small 1- or 2-base-pair deletions that can characterize MSI in endometrial cancer.40 Indeed, an NGS-first strategy41 in endometrial cancer is emerging as potentially more efficient and cost-effective, as it can simultaneously determine MSI status, screen for Lynch syndrome, and provide the complete molecular classification (POLE, TP53) now required for risk stratification. Nevertheless, MMR IHC remains a suitable initial test; however, pathologists interpreting MMR IHC in endometrial cancer patients must be vigilant about identifying and reporting heterogeneous loss of one or more of the MMR proteins, as this pattern of dMMR appears to be more prevalent in endometrial cancers.25
- Prostate Cancer: Limited evidence suggests that the Promega and Bethesda MSI-PCR panels have inferior sensitivity when applied to prostate cancer, with one study showing a sensitivity of only 72.4%.42 In contrast, expanded NGS-based panels that interrogate a larger number of microsatellite loci appear to perform significantly better, with reported sensitivities ranging from 93.1% to 96.6%.42 Thus far, MMR-IHC appears to be an effective screening method for prostate cancer.43,44 Nevertheless, as many prostate cancers are not routinely screened with MMR IHC and/or PCR-based MSI testing, additional studies will be needed to determine which methods provide the most effective initial test and how they compare with one another.
- Other solid tumors: Utilize both MMR IHC and a validated MSI-testing method; any dMMR/MSI-H result can guide tumor-agnostic pembrolizumab eligibility.45–47
Conclusion
Current recommendations suggest that any definitive evidence of MMR deficiency, as determined by either a protein-based assay (IHC) or a DNA-based assay (PCR or NGS), should be considered a positive result for ICI eligibility, regardless of discordant results.2,48 This pragmatic approach, which prioritizes treatment access for all potentially eligible patients, stems from an appreciation of the complexities inherent in cancer biomarker testing. Nevertheless, all discordant results should be reviewed, ideally in a molecular tumor board setting, to exclude resolvable causes like technical artifacts or the effects of neoadjuvant therapy before a final treatment decision is made.
Over the past two decades, the field has moved away from the assumption that phenotypic (IHC) and genotypic (PCR/NGS) assays are interchangeable. Indeed, recent studies confirm that each test can identify dMMR cases that the other misses.5,24 Instead, these tests are now understood as complementary diagnostic tools, as each provides distinct biological information and has unique weaknesses that different methodologies can often compensate for; however, this creates a clinical and economic dilemma. While using multiple tests can identify the greatest number of patients who may benefit from ICI therapy, this comes at a higher financial cost. For instance, a recent study estimated that over 1,100 patients would require NGS-based MSI testing to identify one MSI-H case that IHC would miss. In contrast, nearly 500 patients would require IHC to identify one dMMR case missed by NGS.5 Similarly, these numbers were comparable between MMR IHC and PCR-based MSI testing24; therefore, as testing algorithms are developed, those who make them must strike a balance between minimizing cost and maximizing the identification of truly microsatellite-deficient cases. (See Figure 1 for an example workflow.) The future of MMR/MSI testing is shifting beyond the search for a single "best test" toward the development of tumor-specific, integrated diagnostic algorithms that balance cost with the critical need to maximize the identification of all truly microsatellite-deficient tumors.
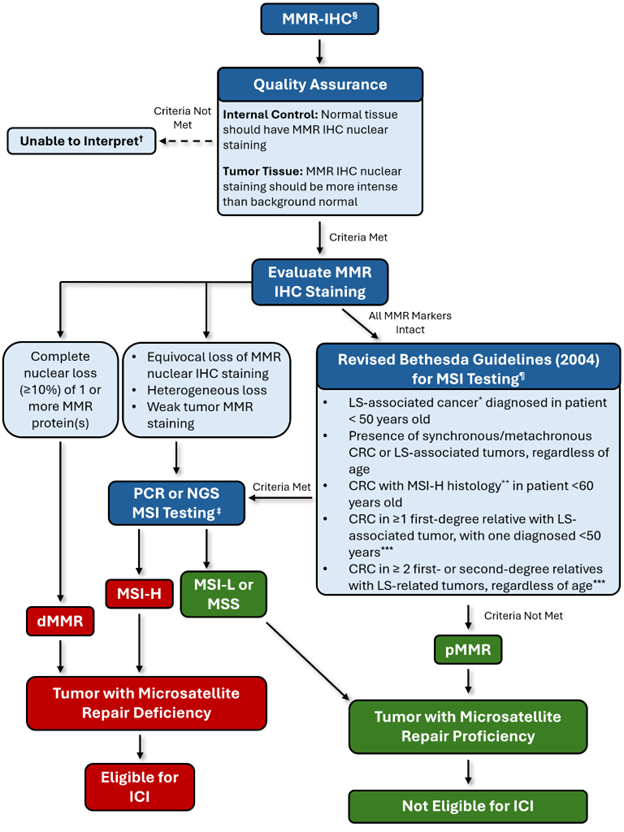
Figure 1: Example workflow that provides high sensitivity for detecting microsatellite repair deficiency while minimizing cost and potential delays in results. § Testing must be performed on an adequate sample, which has at least 50 to 100 viable invasive cancerous cells present and some background stroma/mucosa, which serves as a positive internal control. Avoid interpreting samples that underwent strong-acid decalcification, as this can destroy MMR antigens. If necessary, EDTA-based decalcification is preferred. The current best practice is to test the initial diagnostic biopsy before any chemotherapy and/or radiation therapy has been given to the patient. Chemoradiation has been shown to induce artificial reduction or loss of MSH6 expression.21,22 † If available, use a different block with cancerous cells present. If no additional block is available, attempt PCR-based or NGS-based MSI testing on the block. * LS-associated cancers include colorectal, endometrial, gastric, ovarian, pancreatic, ureter and renal pelvis, biliary tract, glioblastoma, small bowel, and sebaceous gland adenomas and keratoacanthomas. ** MSI-H histology is defined by the presence of tumor-infiltrating lymphocytes, Crohn’s-like lymphocytic reaction, mucinous and/or signet ring differentiation, or medullary growth pattern. *** The last two criteria must be identified by the treating clinical team, with the responsibility for ordering MSI testing likely falling on the medical oncologist. ‡ Either PCR-based and/or NGS-based testing can be utilized based on factors such as availability and cost, as it may be more cost-effective to use NGS-based MSI testing that is paired with a panel such as POLE and TP53 in endometrial cancer, and BRAF in colorectal cancer. It is critical that NGS-based microsatellite testing be shown to be equivalent to PCR-based MSI testing in the same tumor types. ¶ Guidelines slightly modified from the original publication.15 Abbreviations Used: CRC: Colorectal Cancer; dMMR: Mismatch Repair Deficient; ICI: Immune Checkpoint Inhibitor; LS: Lynch Syndrome; MMR: Mismatch Repair; MMR-IHC: Mismatch Repair Immunohistochemistry; MSI: Microsatellite Instability; MSI-H: Microsatellite Instability High; MSI-L: Microsatellite Instability Low; MSS: Microsatellite Stable; NGS: Next Generation Sequencing; PCR: Polymerase Chain Reaction; pMMR: Mismatch Repair Proficient.
Future Outlook
The Role of NGS: NGS-based assays hold promise as a single, comprehensive test for MMR status. A well-designed NGS panel can simultaneously assess thousands of microsatellite loci for instability, calculate the TMB, and sequence the coding regions of all relevant MMR genes. This integrated approach could resolve many discrepancies within a single workflow. In addition, NGS technologies, such as long-read sequencing paired with methylation detection, can simultaneously identify MLH1 promoter hypermethylation and detect large structural variants, deep intronic pathogenic variants, and promoter mutations in the MMR genes; however, significant barriers remain, including higher cost, longer turnaround times, and the critical need for cancer-type-specific validation.
Liquid biopsy: The analysis of circulating tumor DNA (ctDNA) in a blood sample to assess MSI status is an emerging and promising noninvasive technology. Liquid biopsy could help overcome two significant sources of discordance: insufficient tissue from small biopsies and tumor heterogeneity. By sampling DNA shed from all tumor sites, ctDNA analysis may provide a more representative assessment of the overall tumor MSI status. Plasma ctDNA-based MSI assessment has been validated primarily in advanced GI malignancies; however, sensitivity varies with ctDNA shedding and can be limited in low-shedding settings (eg, localized prostate cancer and primary CNS tumors), and it may miss MSI-H subclones present at low fractional abundance in circulation.49–52 Recent findings from the Rome Trial suggest that ctDNA-based liquid biopsies may identify patients with occasional MSI-H tumors who are missed by MMR IHC and NGS testing of the primary tumor.53 Moreover, by testing patients during treatment, liquid biopsies may identify cases in which the cancer evolves from an MSS to an MSI-H phenotype53; however, this technology remains under development, and its sensitivity and specificity relative to other microsatellite repair testing modalities require further validation.
References
- Wang C, Zhang L, Vakiani E, Shia J. Detecting mismatch repair deficiency in solid neoplasms: immunohistochemistry, microsatellite instability, or both? Mod Pathol Off J U S Can Acad Pathol Inc. 2022;35(11):1515-1528.
- Bartley AN, Mills AM, Konnick E, et al. Mismatch Repair and Microsatellite Instability Testing for Immune Checkpoint Inhibitor Therapy: Guideline From the College of American Pathologists in Collaboration With the Association for Molecular Pathology and Fight Colorectal Cancer. Arch Pathol Lab Med. 2022;146(10):1194-1210.
- Ukkola I, Nummela P, Pasanen A, et al. Detection of microsatellite instability with Idylla MSI assay in colorectal and endometrial cancer. Virchows Arch Int J Pathol. 2021;479(3):471-479.
- Yamamoto H, Watanabe Y, Arai H, Umemoto K, Tateishi K, Sunakawa Y. Microsatellite instability: A 2024 update. Cancer Sci. 2024;115(6):1738-1748.
- Ali-Fehmi R, Krause HB, Morris RT, et al. Analysis of Concordance Between Next-Generation Sequencing Assessment of Microsatellite Instability and Immunohistochemistry-Mismatch Repair From Solid Tumors. JCO Precis Oncol. 2024;8:e2300648.
- Bartels S, Grote I, Wagner M, et al. Concordance in detection of microsatellite instability by PCR and NGS in routinely processed tumor specimens of several cancer types. Cancer Med. 2023;12(16):16707-16715.
- Shimozaki K, Hayashi H, Tanishima S, et al. Concordance analysis of microsatellite instability status between polymerase chain reaction based testing and next generation sequencing for solid tumors. Sci Rep. 2021;11(1):20003.
- Ionov Y, Peinado MA, Malkhosyan S, Shibata D, Perucho M. Ubiquitous somatic mutations in simple repeated sequences reveal a new mechanism for colonic carcinogenesis. Nature. 1993;363(6429):558-561.
- Peltomäki P, Lothe RA, Aaltonen LA, et al. Microsatellite instability is associated with tumors that characterize the hereditary non-polyposis colorectal carcinoma syndrome. Cancer Res. 1993;53(24):5853-5855.
- Aaltonen LA, Peltomäki P, Leach FS, et al. Clues to the pathogenesis of familial colorectal cancer. Science. 1993;260(5109):812-816.
- Boland CR, Thibodeau SN, Hamilton SR, et al. A National Cancer Institute Workshop on Microsatellite Instability for cancer detection and familial predisposition: development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res. 1998;58(22):5248-5257.
- Bartley AN, Luthra R, Saraiya DS, Urbauer DL, Broaddus RR. Identification of cancer patients with Lynch syndrome: clinically significant discordances and problems in tissue-based mismatch repair testing. Cancer Prev Res Phila Pa. 2012;5(2):320-327.
- Lindor NM, Burgart LJ, Leontovich O, et al. Immunohistochemistry versus microsatellite instability testing in phenotyping colorectal tumors. J Clin Oncol Off J Am Soc Clin Oncol. 2002;20(4):1043-1048.
- Hatch SB, Lightfoot HM, Garwacki CP, et al. Microsatellite instability testing in colorectal carcinoma: choice of markers affects sensitivity of detection of mismatch repair-deficient tumors. Clin Cancer Res Off J Am Assoc Cancer Res. 2005;11(6):2180-2187.
- Umar A, Boland CR, Terdiman JP, et al. Revised Bethesda Guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite instability. J Natl Cancer Inst. 2004;96(4):261-268.
- Cohen R, Hain E, Buhard O, et al. Association of Primary Resistance to Immune Checkpoint Inhibitors in Metastatic Colorectal Cancer With Misdiagnosis of Microsatellite Instability or Mismatch Repair Deficiency Status. JAMA Oncol. 2019;5(4):551-555.
- Chen J, Yan Q, Sun J, et al. Microsatellite Status Detection of Colorectal Cancer: Evaluation of Inconsistency between PCR and IHC. J Cancer. 2023;14(7):1132-1140.
- Kiss A, Nádorvári ML, Kulka J, et al. Overview of a comparative analysis of microsatellite instability and standard mismatch repair protein-deficiency tests in a large cancer cohort. Pathol Heidelb Ger. 2024;45(Suppl 1):63-66.
- Jaffrelot M, Farés N, Brunac AC, et al. An unusual phenotype occurs in 15% of mismatch repair-deficient tumors and is associated with non-colorectal cancers and genetic syndromes. Mod Pathol Off J U S Can Acad Pathol Inc. 2022;35(3):427-437.
- Evrard C, Tachon G, Randrian V, Karayan-Tapon L, Tougeron D. Microsatellite Instability: Diagnosis, Heterogeneity, Discordance, and Clinical Impact in Colorectal Cancer. Cancers. 2019;11(10):1567.
- Kuan SF, Ren B, Brand R, Dudley B, Pai RK. Neoadjuvant therapy in microsatellite-stable colorectal carcinoma induces concomitant loss of MSH6 and Ki-67 expression. Hum Pathol. 2017;63:33-39.
- Bao F, Panarelli NC, Rennert H, Sherr DL, Yantiss RK. Neoadjuvant therapy induces loss of MSH6 expression in colorectal carcinoma. Am J Surg Pathol. 2010;34(12):1798-1804.
- Orr C, Wang C, Firat C, et al. Primary Clonal Loss of Mismatch Repair Protein on Immunohistochemistry: A Pattern of Abnormality That Warrants Genetic Workup. JCO Precis Oncol. 2022;6:e2200111.
- Ward J, Neff J. Discordant Mismatch Repair Analysis: Impact on Lynch Syndrome Screening and Checkpoint Inhibitor Response. In: Journal of Molecular Diagnostics. Vol 26. 2024:S101-S102.
- Riedinger CJ, Esnakula A, Haight PJ, et al. Characterization of mismatch-repair/microsatellite instability-discordant endometrial cancers. Cancer. 2024;130(3):385-399.
- McCarthy AJ, Capo-Chichi JM, Spence T, et al. Heterogenous loss of mismatch repair (MMR) protein expression: a challenge for immunohistochemical interpretation and microsatellite instability (MSI) evaluation. J Pathol Clin Res. 2019;5(2):115-129.
- Wang T, Stadler ZK, Zhang L, et al. Immunohistochemical null-phenotype for mismatch repair proteins in colonic carcinoma associated with concurrent MLH1 hypermethylation and MSH2 somatic mutations. Fam Cancer. 2018;17(2):225-228.
- Galant N, Krawczyk P, Monist M, et al. Molecular Classification of Endometrial Cancer and Its Impact on Therapy Selection. Int J Mol Sci. 2024;25(11):5893.
- van der Werf-’t Lam AS, Terlouw D, Tops CM, et al. Discordant Staining Patterns and Microsatellite Results in Tumors of MSH6 Pathogenic Variant Carriers. Mod Pathol Off J U S Can Acad Pathol Inc. 2023;36(9):100240.
- Awosika JA, Gulley JL, Pastor DM. Deficient Mismatch Repair and Microsatellite Instability in Solid Tumors. Int J Mol Sci. 2025;26(9):4394.
- Nádorvári ML, Lotz G, Kulka J, Kiss A, Tímár J. Microsatellite instability and mismatch repair protein deficiency: equal predictive markers? Pathol Oncol Res. 2024;30:1611719.
- Chen W, Swanson BJ, Frankel WL. Molecular genetics of microsatellite-unstable colorectal cancer for pathologists. Diagn Pathol. 2017;12(1):24.
- Geurts BS, Zeverijn LJ, van Berge Henegouwen JM, et al. Characterization of discordance between mismatch repair deficiency and microsatellite instability testing may prevent inappropriate treatment with immunotherapy. J Pathol. 2024;263(3):288-299.
- Martínez-Roca A, Giner-Calabuig M, Murcia O, et al. Lynch-like Syndrome: Potential Mechanisms and Management. Cancers. 2022;14(5):1115.
- Ascrizzi S, Arillotta GM, Grillone K, et al. Lynch Syndrome Biopathology and Treatment: The Potential Role of microRNAs in Clinical Practice. Cancers. 2023;15(15):3930.
- Kaplan AR, Glazer PM. Impact of hypoxia on DNA repair and genome integrity. Mutagenesis. 2020;35(1):61-68.
- Grither WR, Hagemann IS, Powell MA, Mullen MM. Attack of the clones: Unveiling subclonal/heterogenous mismatch repair/microsatellite instability status in endometrial cancer. Cancer. 2024;130(3):339-341.
- Bou Farhat E, Adib E, Daou M, et al. Benchmarking mismatch repair testing for patients with cancer receiving immunotherapy. Cancer Cell. 2024;42(1):6-7.
- Sowter P, Gallon R, Hayes C, et al. Detection of Mismatch Repair Deficiency in Endometrial Cancer: Assessment of IHC, Fragment Length Analysis, and Amplicon Sequencing Based MSI Testing. Cancers. 2024;16(23):3970.
- Zhou B, Wang Y, Ding L, et al. A novel algorithm for the detection of microsatellite instability in endometrial cancer using next‑generation sequencing data. Oncol Lett. 2025;29(2):86.
- Rodriguez IV, Strickland S, Wells D, et al. Adoption of Universal Testing in Endometrial Cancers for Microsatellite Instability Using Next-Generation Sequencing. JCO Precis Oncol. 2023;7:e2300033.
- Hempelmann JA, Lockwood CM, Konnick EQ, et al. Microsatellite instability in prostate cancer by PCR or next-generation sequencing. J Immunother Cancer. 2018;6(1):29.
- Yang RK, Chen H, Roy-Chowdhuri S, et al. Clinical Testing for Mismatch Repair in Neoplasms Using Multiple Laboratory Methods. Cancers. 2022;14(19):4550.
- McNevin CS, Keogh A, Mohammed Nur M, et al. Prevalence of Mismatch Repair Deficiency in Primary Prostate Cancer in a Large Prospective Cohort. Clin Cancer Res. 2025;31(9):1746-1753.
- Le DT, Uram JN, Wang H, et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N Engl J Med. 2015;372(26):2509-2520.
- André T, Shiu KK, Kim TW, et al. Pembrolizumab in Microsatellite-Instability-High Advanced Colorectal Cancer. N Engl J Med. 2020;383(23):2207-2218.
- André T, Shiu KK, Kim TW, et al. Pembrolizumab versus chemotherapy in microsatellite instability-high or mismatch repair-deficient metastatic colorectal cancer: 5-year follow-up from the randomized phase III KEYNOTE-177 study. Ann Oncol Off J Eur Soc Med Oncol. 2025;36(3):277-284.
- Vikas P, Messersmith H, Compton C, et al. Mismatch Repair and Microsatellite Instability Testing for Immune Checkpoint Inhibitor Therapy: ASCO Endorsement of College of American Pathologists Guideline. J Clin Oncol. Published online April 1, 2023. Accessed: October 9, 2025.
- Nakamura Y, Okamoto W, Denda T, et al. Clinical Validity of Plasma-Based Genotyping for Microsatellite Instability Assessment in Advanced GI Cancers: SCRUM-Japan GOZILA Substudy. JCO Precis Oncol. 2022;6:e2100383.
- Willis J, Lefterova MI, Artyomenko A, et al. Validation of Microsatellite Instability Detection Using a Comprehensive Plasma-Based Genotyping Panel. Clin Cancer Res Off J Am Assoc Cancer Res. 2019;25(23):7035-7045.
- Lau E, McCoy P, Reeves F, et al. Detection of ctDNA in plasma of patients with clinically localised prostate cancer is associated with rapid disease progression. Genome Med. 2020;12(1):72.
- Huang RSP, Xiao J, Pavlick DC, et al. Circulating Cell-Free DNA Yield and Circulating-Tumor DNA Quantity from Liquid Biopsies of 12 139 Cancer Patients. Clin Chem. 2021;67(11):1554-1566.
- Botticelli A, Cremolini C, Scagnoli S, et al. The Impact of Concordance Between Liquid and Tissue Biopsy for Actionable Mutations: Insights from the ROME Trial. Clin Cancer Res Off J Am Assoc Cancer Res. Published online August 20, 2025. Accessed: October 9, 2025.
- Nyström-Lahti M, Perrera C, Räschle M, et al. Functional analysis of MLH1 mutations linked to hereditary nonpolyposis colon cancer. Genes Chromosomes Cancer. 2002;33(2):160-167.
- Zighelboim I, Powell MA, Babb SA, et al. Epitope-positive truncating MLH1 mutation and loss of PMS2: implications for IHC-directed genetic testing for Lynch syndrome. Fam Cancer. 2009;8(4):501-504.
- Reitsam NG, Märkl B, Dintner S, Waidhauser J, Vlasenko D, Grosser B. Concurrent loss of MLH1, PMS2 and MSH6 immunoexpression in digestive system cancers indicating a widespread dysregulation in DNA repair processes. Front Oncol. 2022;12:1019798.

Jeremy D. Ward, MD, PhD, FCAP, is a molecular and anatomic pathologist at Alberta Precision Laboratories and a Clinical Assistant Professor in the Department of Pathology at the University of Calgary. In his anatomic pathology role, he subspecializes in GI, liver, pancreatic, and transplant pathology. His interests include the application of genomic and proteomic methods for clinical diagnostics and predicting clinical response to targeted therapies and immune checkpoint inhibitors. He is board-certified in anatomic and clinical pathology and has recently completed a fellowship in molecular genetic pathology at Duke University.

Matthew Hiemenz, MD, MS, FCAP, is a senior pathologist and associate medical director at Foundation Medicine. He is board-certified in anatomic and clinical pathology, molecular genetic pathology, and clinical informatics. His interests include assay validation and evidence generation for new assays and biomarkers, biomarker-driven trial design, and the molecular pathology of pediatric solid tumors. He serves on the College of American Pathologists Personalized Health Care Committee.