- Home
- Member Resources
- Articles
- The 'Liquid' Biopsy

Figure 1: (a) Number of publications registered within the PubMed database from 1966 through 2021 matching the search term “liquid biopsy”. Data source: PubMed (https://pubmed.ncbi.nlm.nih.gov/). (b) Relative popularity of the search term “liquid biopsy” in Google search engine from January 2010 through December 2021 compared to the peak popularity in December 2016. The dotted red trendline is over 12 months. Data source: Google Trends (https://www.google.com/trends).
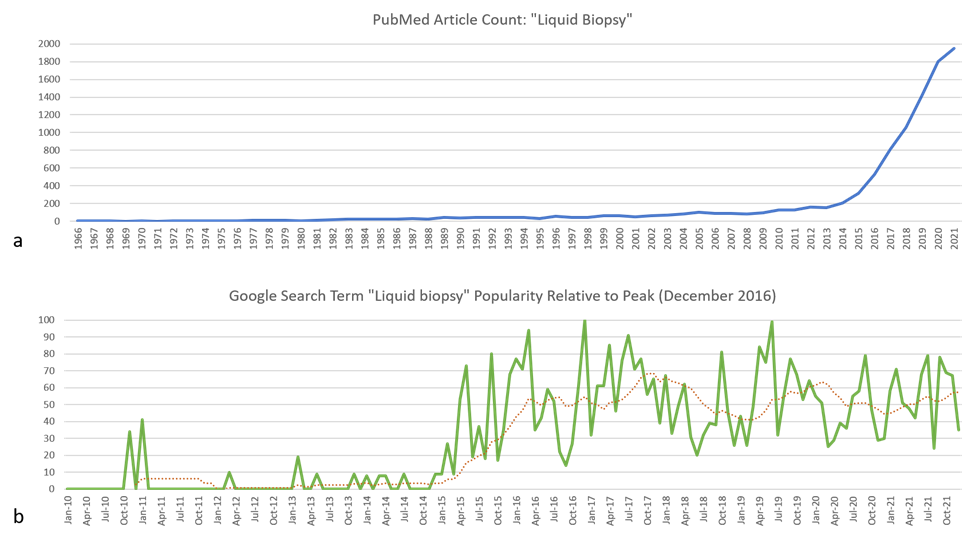
The advent of "precision" medicine has been characterized by improvements in diagnostic, prognostic, and therapeutic approaches. The “liquid biopsy” has recently and rapidly emerged (Figure 1) as a method to minimize the amount of tissue needed for testing, as a less invasive and safer alternative to more traditional procedures. While definitions vary on the precise meaning of this term, it can broadly be thought of as collection of a body fluid sample to test for relevant biomarkers to inform patient management. A “liquid biopsy” does not replace a tissue-based diagnosis, but rather provides alternate sampling for (typically) molecular testing purposes. It is most commonly applied to the collection of peripheral blood for analysis of cell-free circulating tumor deoxyribonucleic acids (DNA).

Figure 2: Tumor cells release cell free DNA/RNA (cfDNA/RNA), circulating tumor cells (CTCs), extracellular vesicles (EVs), and tumor educated platelets (TEPs) via controlled and uncontrolled processes.

The first description of circulating DNA free from cells in human blood was in 1948, but this garnered little attention in the broader scientific community2. In 1977, scientists identified the presence of abnormally high levels of cell-free DNA (cfDNA) in the plasma and serum of cancer patients relative to healthy control patients and this cfDNA was presumed to represent mainly circulating tumor DNA (ctDNA)1,5. Since this original description, other research has found that increased cfDNA generally reflects a multitude of physiologic, such as pregnancy, and pathologic processes, including malignant and benign neoplastic conditions, inflammatory diseases, stroke, trauma, and sepsis2,9. Nucleic acids may be shed into the blood by apoptotic and necrotic cells or by controlled secretion by living cells (Figure 2), usually with peaks in the range of 140-170 base pairs (bp), reflecting their association with nucleosomes2, 3, 9. While cell-free DNA is often used synonymously with circulating tumor DNA, one should remember that circulating non-tumor and non-human nucleic acids may also be present in a sample. For example, the first and still the most widely deployed cfDNA assays are for non-invasive prenatal screening and assays to detect circulating microbial nucleic acids are commercially available. Additional studies examining how non-tumor related processes, such as inflammatory disease states, affect ctDNA in patients are needed12.
While the current focus is predominantly on detection of cfDNA, additional components of a liquid biopsy could include DNA methylation patterns, DNA fragment sizes, ribonucleic acids (RNA), circulating tumor cells, extracellular vesicles or exosomes, and tumor educated platelets. The latter components are mainly of research interest but may have increasing clinical use as more translational research and clinical trials are completed. A review of these elements is beyond the scope of this article and more in-depth discussion can be found elsewhere3, 12.
Technological Considerations

Figure 3: ctDNA in advanced malignancies. Fraction of patients with detectable ctDNA. Error bars represent the 95% bootstrapped confidence interval of the mean (tumor types with <4 samples were excluded from this figure)8.
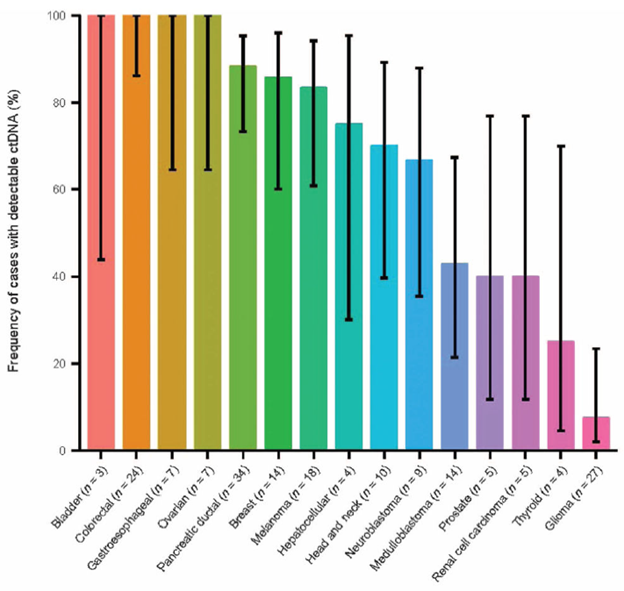
Until the mid-2010s, available technology was not sufficiently sensitive to detect ctDNA for clinical use. Whereas nucleic acids from viruses and other microorganisms comprise comparatively large quantities of foreign nucleic acids, circulating tumor nucleic acid fragments exist at a small fraction of the normal non-tumor cfDNA and are more challenging to differentiate and amplify. Variant allele fractions are generally much lower, often <1%, in liquid biopsies than tissue biopsies and several tumor specific factors affect the rate and quantity of ctDNA shedding into the blood. Generally, tumors with larger volume, higher stage, increased cellular proliferation, and increased vascularity tend to shed more ctDNA 8. Certain tumor types also shed more than others, such as carcinomas versus gliomas (Figure 3)8. In addition, although ctDNA is relatively stable while in circulation, it is removed from circulation by the liver, kidneys, and spleen within hours3, 4, 14. In vitro, it is important to minimize the time between blood draw and separating plasma or serum from other blood elements or utilization of specialized cell stabilizing media to avoid artifacts such as ctDNA dilution by non-tumor genomic DNA or nuclease degradation which may lead to unreliable results13-15. These technical considerations and other advances in pre-analytic processes and purification methods are more likely to lead to successful capture, amplification, and sequencing of ctDNA13. Additionally, although most interest has been directed at blood specimens, assays using other fluids such as urine and CSF have also been deployed2, 4.
Methods that are currently used to detect or measure ctDNA can broadly be divided into two categories: polymerase chain reaction (PCR) based and next-generation sequencing (NGS) based. PCR-based assays generally have a faster turnaround time and are less expensive, but typically can only assess one or a few specific variants at a time and cannot complete more sophisticated analyses such as assignment of tissue of origin or tumor mutation burden. Limits of detection vary between different methodologies but have been described as low as 0.1% allele fraction10. NGS libraries are prepared from plasma DNA using either ligation/hybrid capture or targeted PCR enrichment methods approaches and target larger genomic regions of interest. This permits assessment of a much broader range of possible variants since sequencing, rather than locus-specific genotyping, can detect most variants occurring within regions covered by the assay. NGS approaches therefore have not only the important advantage of being able to achieve much broader variant coverage but can also (depending on assay design) identify additional tumor properties such as tumor mutational burden and microsatellite instability for immunotherapy applications12. Limits of detection down to 0.1% can be achieved for single nucleotide variants and are variably higher for more complex genomic aberrations such as insertions/deletions and copy number variants10. Methods utilizing whole exome/genome sequencing have also been described10. Since variant allele fractions are lower in liquid biopsies than tissue biopsies, regions of interest must be sequenced more deeply than in NGS of primary tumor tissue to ensure assay accuracy. This impacts the practical size and cost of liquid biopsy panels which often cover less of the genome and/or are costlier. In addition, extensive optimization of pre-analytic and analytic processes needs to be done to maximize input sample and reduce PCR and sequencing errors. Compared to simpler PCR-based methods, NGS techniques are more expensive, time consuming, and technically challenging, both at the wet bench and during interpretation.
Advantages and Disadvantages
Assays to detect ctDNA carry several direct advantages over tissue-based sequencing as well as some which are the subject of ongoing investigation. One of the clearest benefits of ctDNA assays is the molecular assessment of lesions in inaccessible or risky locations. Consider, for example, the process involved to biopsy a lung mass. Even when the mass lies in a location that is accessible by interventional radiology or surgical biopsy, there is a risk of potentially life-threatening complications including pneumothorax or hemorrhage versus the very low risk of a peripheral blood draw. Another potential advantage is the greater likelihood that ctDNA represents a greater mixture of subclones in a tumor and any metastases than any single biopsied site, helping to eliminate treatment failures due to intratumoral and intertumoral heterogeneity that may be missed by tissue sequencing 11, 12. Liquid biopsies may also allow more frequent and serial samplings over time to provide greater resolution to the behavior of tumors and their response to therapy (Figure 3)8. These concepts are analogous to testing that is currently done for hematologic malignancies, such as serial BCR::ABL1 fusion transcript monitoring in patients with chronic myelogenous leukemia (CML).
Important disadvantages of liquid biopsy include the need for an initial histologic diagnosis to be obtained by tissue biopsy. Assays for ctDNA do not provide any insight into many tumor properties important for grading and staging and only indirectly suggest overall tumor burden. As previously discussed, some neoplasms are known to release very little or no DNA into circulation, and studies to show how properties of the tumor such as location within an organ or physiologic states such as inflammation affect ctDNA detection. The very low variant frequency in ctDNA may lead to greater risks of both false negatives (from variants below the assay’s limit of detection) and false positives (from the effects of background sequencing error). Assays meant for ctDNA are more technically sensitive but also often more limited in genomic scope than those meant for tissue, which can lead to discrepancies when both types of assays are performed on the same patient. As noted above, inter- and intratumoral heterogeneity may be mitigated by ctDNA assays, but this also presents unique challenges when a patient may have more than one clonal process occurring. For example, imagine a ctDNA assay for a lung cancer patient reveals the BRAF p.V600E variant in the patient’s blood. This variant may belong to the lung cancer or potentially an undiagnosed thyroid carcinoma, colorectal adenocarcinoma, multiple myeloma, melanoma, or even benign nevi. Additionally, the blood itself may carry somatic variants as seen in clonal hematopoiesis which is a recognized confounder that requires significantly greater technical efforts and expertise to obtain reliable results 12. Laboratories performing these assays need to be mindful of appropriate test utilization, the risks of false positives and false negatives, and the potential for "over interpretation" in the clinical context.
Current and Emerging Applications

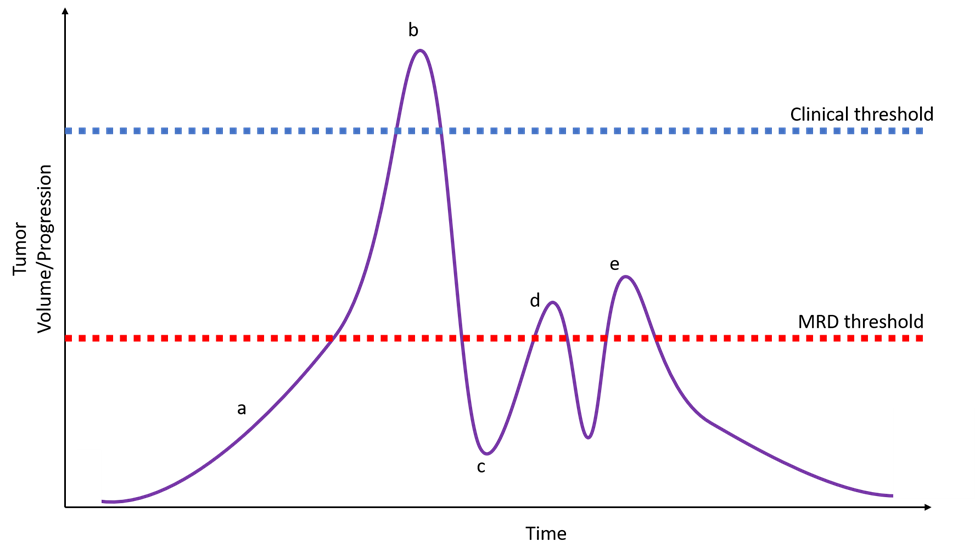
Figure 4: A hypothetical patient with a very early developing cancer that might be detected before clinical presentation via liquid biopsy in a population screening protocol (a). The patient eventually presents with clinically and radiographically apparent disease (b) and initiates treatment tailored to tumor variants identified via tumor sequencing complemented by liquid biopsy. The liquid biopsy identifies relevant variants not seen in the primary tumor allowing the oncologist to start with second-generation targeted therapy. Post-treatment testing via liquid biopsy shows no detectable disease (c). Subsequent liquid biopsy monitoring identifies a low-level recurrence of disease (d) triggering additional treatment. Subclinical recurrence is again detected with identification of a resistance variant (e) and a third-generation targeted therapy is used with a favorable outcome. MRD = measurable residual disease.
The clinical use of the liquid biopsy has significantly increased since 2014, when the first commercially available multigene liquid biopsy platform became available. Initially, use was largely driven by patients and physicians desire to identify targetable variants for off-label drug use or clinical trial enrollment. Several assays are now commercially available and FDA-approved, and some are considered sufficient for the determination of treatment eligibility by insurance companies. For example, in 2016 the FDA approved the cobas® EGFR Mutation Test v2 to determine the eligibility of non-small cell lung cancer patients to receive certain EGFR tyrosine kinase inhibitors7. NGS-based assays have also received FDA approval as companion diagnostics with the Guardant360® CDx assay in 2020 for non-small cell lung cancer and the FoundationOne® Liquid CDx assay in 2021 for non-small cell lung, prostate, ovarian, and breast cancer12. Both assays have also received approval as complementary diagnostic tests for tumor mutation profiling of solid tumors12. Adoption of the liquid biopsy to exclude patients from targeted therapy has seen much slower clinical adoption, mostly due to concerns for false negatives and generally accessible tumor tissue3. At this time, negative liquid biopsy sequencing should be followed up by tissue biopsy sequencing12.
Emerging uses for liquid biopsies (Figure 4) include assessing treatment response, residual disease monitoring and detection of disease recurrence, monitoring for resistance variants, and expanding use to different tumor types; additional discussion can be found elsewhere3, 12, 16.
Future Directions
The expected cancer-specificity of variants makes ctDNA an attractive biomarker for early detection of cancer or population screening (Figure 4), which could have tremendous impact on patient care. However, since early-stage tumors are known to release very little DNA, there are many technical challenges to be overcome. Such applications will need to be carefully and thoughtfully considered to avoid excessive patient suffering and cost due to false positive results or misleading reassurance due to false negative results. In the short term, liquid biopsies may be more helpful to confirm malignancy in patients with already clinically or radiographically apparent lesions and otherwise inaccessible tumor tissue.
Assays combining multiple biomarkers of ctDNA, CTCs, cfRNA, extracellular vesicles, and/or protein biomarkers are active areas of research and may allow greater resolution to liquid biopsies 4, 12. Application of artificial intelligence to aid in the analysis and interpretation of liquid biopsy is also an area of active research 12.
Conclusion
The literature focusing on liquid biopsies is quickly expanding and evolving. Ongoing areas of investigation include pre-analytical processes, factors affecting ctDNA detection rate, and prospective clinical trials and the reader is encouraged to review the included references. We are likely to see continued integration of ctDNA in the clinical care of patients with advanced non-small cell lung cancers with eventual application to a broader swath of tumor types and earlier stage disease. Emerging uses will include disease monitoring at various stages of treatment. Future integration of additional biomarkers may allow more technically and ethically complicated applications such as population screening. There is a great deal of enthusiasm for liquid biopsy and the potential it has to revolutionize cancer care; however, widespread adoption will necessarily need to be tempered by evolving evidence for not just analytical validity but also clinical validity and utility11.
References
- Chakravadhanula M, Tembe W, Legendre C, et al. Detection of an Atypical Teratoid Rhabdoid Brain Tumor Gene Deletion in Circulating Blood Using Next-Generation Sequencing. J Child Neurol 2013;29(9):NP81-NP85.
- Schwarzenbach H, Hoon DSB, Pantel K. Cell-free nucleic acids as biomarkers in cancer patients. Nat Rev Cancer 2011; 11: 426-37.
- Batth IS, Mitra A, Manier S, et al. Circulating tumor markers: harmonizing the yin and yang of CTCs and ctDNA for precision medicine. Ann Oncol 2017;28(3):468-477.
- Best MG, Sol N, Zijl S, et al. Liquid biopsies in patients with diffuse glioma. Acta Neuropathol 2015; 129: 849–865.
- Leon SA, Shapiro B, Sklaroff DM, Yaros MJ. Free DNA in the Serum of Cancer Patients and the Effect of Therapy. Cancer Res 1977 March; 37: 646-650.
- Patel AA, Chandra P. Emerging Concepts on Liquid Biopsy: Circulating Tumor in Cell Free DNA (cfDNA). Short Presentations on Emerging Concepts. College of American Pathologists, 6 Sept 2016. World Wide Web. Accessed 12 August 2017.
- "cobas® EGFR Mutation Test v2 - P150047". Medical Devices. U.S. Food and Drug Administration, 9 Sept 2016. World Wide Web. Accessed 20 Sept 2017.
- Bettegowda C, Sausen M, Leary RJ, et al. Detection of Circulating Tumor DNA in Early- and Late-Stage Human Malignancies. Sci Transl Med 2014 Feb 19;6(224):224ra24/
- Diaz LA Jr, Bardelli A. Liquid Biopsies: Genotyping Circulating Tumor DNA. J Clin Oncol 2014 Feb 20; 32(6):579-86.
- Pessoa LS, Heringer M, Ferrer VP. ctDNA as a cancer biomarker: A broad overview. Crit Rev Oncol Hematol. 2020 Nov;155:103109.
- Merker JD, Oxnard GR, Compton C, et al. Circulating Tumor DNA Analysis in Patients with Cancer: American Society of Clinical Oncology and College of American Pathologists Joint Review. J Clin Oncol. 2018 Jun 1;36(16):1631-1641.
- Ignatiadis M, Sledge GW, Jeffrey SS. Liquid biopsy enters the clinic - implementation issues and future challenges. Nat Rev Clin Oncol. 2021 May;18(5):297-312.
- Meddeb R, Al Amir Dache Z, Thezenas S, Otandault A, Tanos R, et al. Quantifying circulating cell-free DNA in humans. Sci Rep. 2019 Mar 26;9(1):5220.
- Elazezy M, Joosse SA. Techniques of using circulating tumor DNA as a liquid biopsy component in cancer management. Comput Struct Biotechnol J. 2018 Oct 9;16:370-378.
- Compton CC, Robb JA, Anderson MW, Berry AB, Birdsong GG, et al. Preanalytics and Precision Pathology: Pathology Practices to Ensure Molecular Integrity of Cancer Patient Biospecimens for Precision Medicine. Arch Pathol Lab Med. 2019 Nov;143(11):1346-1363.
- Pantel K, Alix-Panabières C. Liquid biopsy and minimal residual disease - latest advances and implications for cure. Nat Rev Clin Oncol. 2019 Jul;16(7):409-424.

Damon R. Olson, MD, FCAP, is Molecular Pathology Program Director and Associate Medical Director of Anatomic Pathology at Children's Minnesota. He is board-certified in Molecular Genetic Pathology, Pediatric Pathology, and Anatomic and Clinical Pathology. He serves on the Personalized Health Care Committee of the College of American Pathologists and the Education Committee of the Society of Pediatric Pathology.