Clinical Summary
A 67-year-old man presents with hematuria. An abdominal computed tomography (CT) scan reveals a 9 cm mass in the upper pole of the left kidney. A radical nephrectomy is performed. Gross examination reveals a 9.3 x 9.0 x 6.5 cm circumscribed, unencapsulated, golden-yellow, solid and cystic mass with areas of hemorrhage. The tumor appears to invade into the hilar fat. Surgical margins, including distal ureter and renal vein, are negative for tumor. The adrenal gland is unremarkable.
Master List of Diagnoses
- Adrenocortical neoplasm
- Chromophobe renal cell carcinoma
- Clear cell papillary renal cell carcinoma
- Clear cell renal cell carcinoma
- MiT family translocation renal cell carcinoma
- Papillary renal cell carcinoma
Archive Case and Diagnosis
This case first appeared as Performance Improvement Program in Surgical Pathology (PIP) 2018, Case 10, and is clear cell renal cell carcinoma of the kidney.
Criteria for Diagnosis and Comments
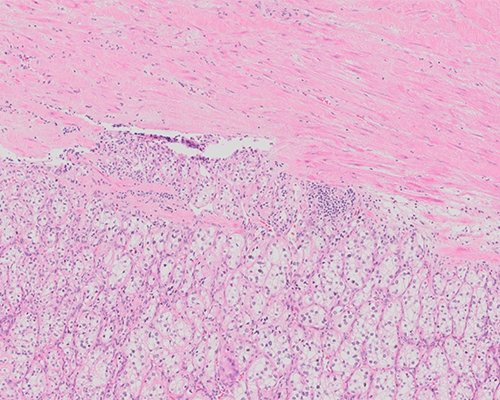
Histologic sections show a circumscribed tumor composed of cells arranged in nested and trabecular patterns intimately associated with numerous small, thin-walled vessels. The tumor cells have clear cytoplasm and distinct cell membranes. The nuclei are round with evenly distributed chromatin and small nucleoli visible at 400x magnification. There are no areas of necrosis, cystic change, or sarcomatoid change. Based on the morphology, this is a clear cell renal cell carcinoma (RCC-CC) with a World Health Organization (WHO)/International Society of Urological Pathology (ISUP) grade of 2.
RCC-CC accounts for 65% to 70% of all cases of renal cell carcinoma. It originates from epithelial cells lining the proximal tubule and is the most aggressive subtype among the common subtypes of renal cell carcinoma. Although any age group may be affected, it usually presents in patients over 40 years old and has a slight male preponderance. Most are discovered incidentally on imaging studies. Symptoms may include hematuria and flank pain. These tumors have diverse metastatic pathways, including hematogenous and lymphatic spread, which enable them to metastasize to unusual sites throughout the body. The most common site of metastasis is the lung, followed by the liver and bone.
Most RCC-CC occur sporadically, but may be associated with risk factors such as obesity, smoking, and occupational exposures (organic solvents such as trichloroethylene, metals such as cadmium, and certain herbicides) or in various familial settings such as von Hippel Lindau syndrome, Cowden syndrome, Birt-Hogg-Dube syndrome, tuberous sclerosis, and succinate dehydrogenase-deficient renal cell carcinoma. Inactivation of the von Hippel-Lindau tumor suppressor gene (VHL) was originally identified in association with the von Hippel-Lindau syndrome, a familial syndrome that predisposes one to RCC-CC. The VHL gene is a 2-hit tumor suppressor gene on the short arm of chromosome 3 (3p25.3). Subsequently, the VHL was found to be inactivated in approximately 90% of sporadic RCC-CC. Loss of function of the von Hippel-Lindau protein causes tumor initiation, progression, and metastasis.
Additional genes have been discovered in this region of chromosome 3 that are also associated with RCC-CC: PBRM1 (mutated in 45%), BAP1 (mutated in 10% to 15%), and SETD2 (mutated in 10% to 15%). All of these genes are 2-hit tumor suppressor genes. Typically, one allele of the VHL gene is inactivated through mutation, and the second is deleted as part of a large deletion of chromosome 3p. Deletions of chromosome 3p are identified in approximately 90% of sporadic RCC-CC. BAP1 and PBRM1 mutations are usually mutually exclusive and are associated with different histologic features and outcomes. PBRM1-mutant tumors may be of high- or low-grade and less frequently show necrosis. BAP1-mutant tumors typically are high-grade and show coagulative necrosis; they also have a higher hazard ratio for death than PBRM1–mutant tumors. An immunohistochemical test has been developed to determine BAP1 loss.
Several additional genetic characteristics have also been associated with a poor prognosis, including losses on chromosomes 14q, 4p, and 9p. Advances in molecular genetics have contributed to a better understanding of RCC-CC and the prediction of the biology and prognosis of individual tumors. A single tumor, however, may harbor several different genetic mutations and this genetic heterogeneity is one of the major obstacles in developing oncologic therapies against renal cancers.
Grossly, RCC-CCs are usually unilateral, solitary, cortical tumors, although bilaterality and multicentricity may be seen in hereditary forms. The cut surface shows a characteristic golden yellow color imparted by cholesterol and lipids within the tumor cells. Other appearances include gray-white areas of fibrosis, hemorrhage, cystic change, and necrosis. It is typically well-circumscribed, surrounded by a fibrous pseudocapsule, and has an expansile growth pattern.
The microscopic appearance of RCC-CC is also variable. Common architectural patterns include nests, alveoli, cysts, and sheets. Papillary architecture may be seen focally but typically does not represent a significant portion of the tumor. Prominence of papillary architecture should raise the diagnostic possibility of MiT family translocation associated renal cell carcinoma (RCC-MiT), or clear cell papillary renal cell carcinoma (RCC-CCP). Tumor cells are typically surrounded by delicate fibrovascular septations. Tumor cells have distinct cell membranes and clear cytoplasm due to the loss of cytoplasmic lipids during histologic processing. In some cases, cells may have granular eosinophilic cytoplasm, raising the diagnostic consideration of a chromophobe renal cell carcinoma (RCC-CH), but this feature is usually seen in higher grade tumors or adjacent to areas of hemorrhage or necrosis. The nuclei may vary in size and shape, from small round nuclei to enlarged and irregular nuclei and are an important parameter to assess grading. Calcification, fibromyxoid areas, and ossification may also be seen, but have no known prognostic significance. These tumors typically incite very little inflammatory response. RCC-CC is typically positive for epithelial markers (AE1/AE3, CAM5.2, EMA) renal epithelial markers (PAX8, PAX2, RCC), vimentin, and CD10. Carbonic anhydrase IX (CAIX) is typically positive in a complete membranous distribution, although its expression may be reduced in high-grade tumors. This tumor is typically negative for CK7, CD117, AMACR, TFE3, Melan-A, and Inhibin.
Pathologic stage is the most important prognostic indicator for RCC-CC. After stage and tumor grade, the presence of necrosis and sarcomatoid/rhabdoid change are also prognostic parameters. The WHO/ISUP 4-tiered grading system is used to grade RCC-CC. Grades 1 to 3 are based on the degree of nucleolar prominence, and grade 4 is assigned to those tumors showing any degree of sarcomatoid or rhabdoid differentiation, as well as tumors with marked nuclear pleomorphism or tumor giant cells. Grading should be assigned on the basis of the single high-power field with the highest grade. Although the current grading system is based only on nuclear appearance, some authors have suggested that tumor necrosis also be incorporated into the grading system, as it has prognostic significance. Regardless, the amount of necrosis should be quantified and reported. Greater than 10% tumor necrosis is considered a significant adverse prognostic indicator (> 20% for TNM stage 1 and 2 tumors). A variety of molecular markers have been proposed as prognostic indicators; their utility is limited in daily practice, however, due to limitations in specificity, predictive accuracy, and tumor heterogeneity.
The current treatment of RCC-CC is surgery with either radical nephrectomy or partial nephrectomy, depending on the size and location of the tumor. Due to advancing surgical techniques and the discovery of tumors at smaller sizes, there is a shift toward partial nephrectomy in order to preserve renal function. Chemotherapy and radiation therapy may be utilized for advanced disease, but do not typically yield a good result. Immunotherapies similarly have not yielded positive results. Newer targeted therapies are being investigated.
The differential diagnosis of RCC-CC includes other types of renal cell carcinomas (RCC) as well as neoplasms that may show clear cell features. Adequate sampling of the tumor for the characteristic morphologic features and, in some cases, immunohistochemistry can be helpful in making the diagnosis.
Adrenocortical neoplasms may have tumor cells with clear cytoplasm, however these tumors have a vastly different immunophenotype than RCC, as they are negative for EMA and cytokeratins and positive for inhibin and Melan-A.
Of the subtypes of RCCs, those that may be confused with RCC-CC include clear cell papillary RCC, chromophobe RCC, papillary RCC, and MiT family translocation RCC. Attention to morphology and an appropriate immunohistochemical panel will usually resolve any confusion. RCC-CC may show papillary or pseudopapillary architecture, but this should be a minor component of the tumor. The papillae of papillary RCC typically have foamy histiocytes within well-formed fibrovascular stalks and stroma. RCC-CC may show eosinophilic granular cytoplasm, which may lead to confusion with chromophobe RCC. Similarly, chromophobe RCC may show areas where the cells have clear cytoplasm. Chromophobe RCC, however, shows the characteristic raisinoid appearance of the nuclei with perinuclear clearing, prominent plant-like cell borders, and usually incomplete fibrovascular septations.
MiT family translocation RCC may have a combination of papillary architecture and clear cell morphology and typically have a high nuclear grade, all features that may be seen in RCC-CC. MiT family translocation RCC has a characteristic immunoprofile, however, and is positive for TFE3 or TFEB, may express melanoma-associated markers (HMB-45 and Melan-A), and some are positive for cathepsin K; these tumors are typically negative for EMA and cytokeratins. Clear cell papillary RCC typically has prominent papillary architecture with clear cell morphology. These tumors usually have a low nuclear grade and a characteristic pattern in which the nuclei are arranged in a linear manner away from the basement membrane. Typical RCC-CC does not have a prominent papillary architecture. The table below lists the characteristic immunohistochemical stains that are positive in the most common subtypes of RCC (Table 1).
Table 1. Typical immunohistochemical features of RCC subtypes
RCC Subtype |
CK7 |
AMACR |
CD10 |
CAIX |
CD117 |
TFE3/TFEB |
EMA |
AE1/AE3 |
Vimentin |
Clear cell |
- |
- |
+ |
+* |
- |
- |
+ |
+ |
+ |
Papillary |
+ |
+ |
+ |
- |
- |
- |
+ |
+ |
+ |
Chromophobe |
+ |
- |
- |
- |
+ |
- |
+ |
+ |
- |
MiT family translocation |
- |
+ |
+/- |
- |
- |
+ |
- |
- |
-/+ |
Clear cell papillary |
+ |
- |
- |
+* |
- |
- |
+ |
+ |
+ |
*Clear cell RCC shows complete membranous staining with CAIX whereas clear cell papillary RCC shows a basolateral staining pattern.
Supplementary Questions
- Which of the following histologic features is an indicator of poor prognosis in clear cell renal cell carcinoma?
- Eosinophilic granular cytoplasm
- Osseous metaplasia
- Papillary formations
- Sarcomatoid differentiation
- Small round nuclei with inconspicuous nucleoli
- Which of the following immunohistochemical markers is typically positive in clear cell renal cell carcinoma?
- AMACR
- CAIX
- CD117
- CK7
- TFE3
- Which of the following chromosomal abnormalities may be seen in clear cell renal cell carcinoma?
- Activating mutations of MET oncogene on 7q31
- Deletions of chromosome 9p
- Loss of chromosome 3p
- Mutations of the p53 gene
- Trisomy of chromosomes 7 and 17 and loss of chromosome Y
References
- Amin MB, Amin MB, Tamboli P et al. Prognostic impact of histologic subtyping of adult renal epithelial neoplasms: an experience of 405 cases. Am J Surg Pathol. 2002;26(3):281-291.
- Brugarolas J. Molecular genetics of clear-cell renal cell carcinoma. J Clin Oncol. 2014;32(18):1968-1976.
- Delahunt B, Cheville JC, Martignoni G, et al. The international society of urologic pathology (ISUP) grading system for renal cell carcinoma and other prognostic parameters. Am J Surg Pathol. 2013;37(10):1490-1504.
- Delahunt B, McKenney JK, Lohse CM, et al. A novel grading system for clear cell renal cell carcinoma incorporating tumor necrosis. Am J Surg Pathol. 2013;37(3):311-322.
- Eble JN, Sauter G, Epstein JI, et al. eds. WHO Classification of Tumours of the Urinary System and Male Genital Organs. 2nd Ed. IARC Press;2016:1:31-1:40.
- Khor LY, Dhakal HP, Jia X, et al. Tumor necrosis adds prognostically significant information to grade in clear cell renal cell carcinoma: A study of 842 consecutive cases from a single institution. Am J Surg Pathol. 2016;40(9):1224-1231.
- Przybycin CG, McKenney JK, Reynolds JP, et al. Rhabdoid differentiation is associated with aggressive behavior in renal cell carcinoma: a clinicopathologic analysis of 76 cases with clinical follow-up. Am J Surg Pathol. 2014;38(9):1260-1265.
- Simmons MN Weight CJ, Gill IS. Laparoscopic radical versus partial nephrectomy for tumors > 4 cm: intermediate-term oncologic and functional outcomes. Urology. 2009;73(5):1077-1082.
- Skinnider BF, Amin MB. An immunohistochemical approach to the differential diagnosis of renal tumors. Semin Diagn Pathol. 2005;22(1):51-68.
- Stukalin I, Alimohamed N, Heng DY. Contemporary treatment of metastatic renal cell carcinoma. Oncol Rev. 2016;10(1):295.
- Truong L, Shen S. Immunohistochemical diagnosis of renal neoplasms. Arch Pathol Lab Med. 2011;135:92-109.
Author
Danielle E. Westfall, MD
Surgical Pathology Committee
TOPA Diagnostics
Thousand Oaks, CA
Answer Key
- Sarcomatoid differentiation (d)
- CAIX (b)
- Loss of chromosome 3p (c)