- Home
- Member Resources
- Pathology Case Challenge
- Pancreas
Clinical Summary
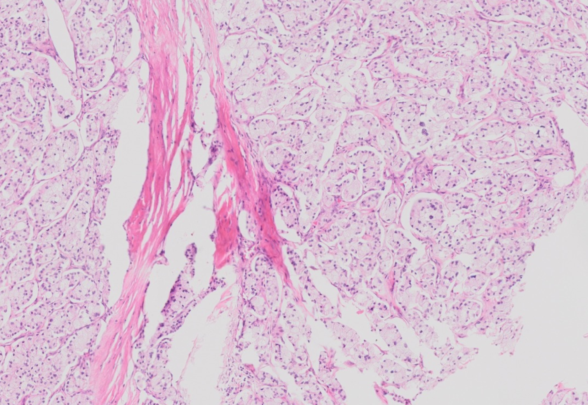
A 70-year-old man presents with worsening abdominal discomfort of several months’ duration. Radiologic studies reveal a 6 cm mass in the head of the pancreas. A partial pancreatectomy with duodenectomy is performed. The resected specimen shows a well-circumscribed, firm mass measuring 6.0 x 5.5 x 3.2 cm in the head of the pancreas. The cut surface is bright yellow with a vaguely lobular appearance. By immunohistochemical studies, the lesional cells are positive for cytokeratin AE1/3, chromogranin, and synaptophysin.
Master List of Diagnoses
- Acinar cell carcinoma
- Metastatic renal cell carcinoma
- Pancreatic adenocarcinoma
- Pancreatic neuroendocrine tumor, clear cell variant
- Solid pseudopapillary neoplasm
Archive Case and Diagnosis
This case first appeared as Performance Improvement Program in Surgical Pathology (PIP) 2018, Case 38, and is a pancreatic neuroendocrine tumor, clear cell variant in the pancreas.
Criteria for Diagnosis and Comments
The histologic sections show a fairly well-defined neoplasm separated from the adjacent pancreatic parenchyma and soft tissue by a rim of fibrous tissue. The neoplastic cells are arranged in nests and trabeculae separated by broad zones of dense hyalinized stroma. The cells have abundant finely vacuolated microvesicular cytoplasm resulting from accumulation of lipid droplets that may scallop the nucleus. The nuclei are round to oval with stippled chromatin and show minimal atypia. Mitotic activity is low at less than 1 per 10 high-power fields (HPFs). By immunohistochemical stains, the neoplastic cells are positive for cytokeratin AE1/3, cytokeratin CAM5.2, synaptophysin, chromogranin, and CD56, and have a low Ki-67 labeling index of approximately 1%. The overall findings are consistent with clear cell variant of well-differentiated neuroendocrine tumor of the pancreas (WHO grade 1).
Pancreatic neuroendocrine neoplasms (PanNENs) account for 2% to 5% of all primary pancreatic tumors and include malignant well-differentiated neuroendocrine tumors (PanNETs) and poorly differentiated neuroendocrine carcinomas (PanNECs). These neoplasms are characterized by significant neuroendocrine differentiation with expression of synaptophysin and usually chromogranin A. They occur with equal frequency in men and women, most frequently between 30 and 60 years of age. Tumors that are associated with genetic syndromes (eg, multiple endocrine neoplasia type 1, and von Hippel-Lindau syndrome) tend to occur at a younger age. Tumors associated with a clinical syndrome due to hypersecretion of a hormone (insulinoma, glucagonoma, etc) are classified as “functional PanNETs,” which accounted for 60% to 85% of all PanNENs in the past. However, recent data show that nonfunctioning PanNETs now account for more than 60%, likely due to enhanced diagnostic imaging. The head and tail of the pancreas are common sites of involvement, with approximately two-thirds of surgically resected nonfunctioning PanNETS occurring in the head of the pancreas.
PanNETs are generally slow growing, solitary, and well-demarcated yellow to pink soft lesions ranging in size from 2 to 12 cm. Larger tumors can have foci of intratumoral hemorrhage. The neoplastic cells are arranged as pseudoglands, nests, or trabeculae embedded in fibrotic stroma. The cells are relatively uniform with cytoplasm that is often finely granular and eosinophilic, a round to oval nucleus with characteristic coarse chromatin arranged in “salt and pepper” clumps, and a prominent nucleolus. A number of morphologic variants have been described. The oncocytic variant has cells with abundant granular eosinophilic cytoplasm. The pleomorphic variant has large irregular and atypical nuclei. The clear cell (lipid-rich) variant shows innumerable vacuoles in the cytoplasm as a result of lipid accumulation; this variant has been reported more commonly in patients with von Hippel-Lindau syndrome. Although not always readily apparent, mitoses are a critical component of tumor grading in PanNETs.
The recent WHO classification (2017) classifies PanNETs into grade 1 (mitoses less than 2 per 10 HPFs and Ki-67 index less than 3%); grade 2 (mitoses 2 to 20 per 10 HPFs or Ki-67 index 3% to 20%); and grade 3 (mitoses more than 20 per 10 HPFs or Ki-67 index greater than 20%). This new tumor category of PanNET grade 3 is reserved for tumors that retain a well-differentiated histologic pattern but show a Ki-67 proliferation of greater than 20%, distinguishing it from PanNEC.
PanNEC is a poorly differentiated high-grade neoplasm (G3) composed of highly atypical small or large cells expressing general markers of neuroendocrine differentiation and rarely hormones, and lacking expression of exocrine enzyme markers such as trypsin and chymotrypsin. The TNM classification for these tumors follows the criteria for classifying ductal adenocarcinoma.
Acinar cell carcinoma is characterized by large cells in acinar formations, granular apical cytoplasm, and basally oriented nuclei with a characteristic single prominent nucleolus. This neoplasm has a high mitotic rate and shows immunopositivity for trypsin, chymotrypsin, and lipase. Labeling with BCL10 can be also helpful in distinguishing this tumor from PanNET.
Metastatic renal cell carcinoma should be considered in the histologic differential diagnosis of the clear cell variant of PanNET. However, renal cell carcinoma cells are negative for neuroendocrine markers.
Pancreatic adenocarcinoma is an infiltrating lesion composed of variably shaped glands lined by atypical cuboidal to columnar cells with mucin secretion. The mitotic activity is high in comparison to well-differentiated PanNETs.
Solid-pseudopapillary neoplasm, found predominantly in young women, is characterized by a heterogenous pattern of solid, pseudopapillary, hemorrhagic, and necrotic areas. Microscopically, poorly cohesive uniform cells are admixed with delicate vascular structures, histiocytes, and cholesterol crystals. Tumors with a predominantly solid growth pattern can mimic PanNETs. Like pancreatic NETs, they label with CD56 and synaptophysin; however, chromogranin is not expressed. In addition, periodic acid-Schiff (PAS) positive hyaline globules, expression of CD10, and nuclear labeling for β-catenin are characteristic of solid pseudopapillary neoplasms.
Supplementary Questions
- Which of the following is most helpful in differentiating a pancreatic neuroendocrine neoplasm from solid pseudopapillary neoplasm?
- CD56 positivity
- Labeling for BCL10
- Labeling for nuclear β-catenin
- Solid growth pattern
- Synaptophysin positivity
- Based on the 2017 WHO classification of pancreatic neuroendocrine neoplasms, which of the following is true regarding grade 3 pancreatic neuroendocrine tumor (PanNET)?
- Chromogranin positive with a Ki-67 index of >20%
- Poorly differentiated tumors with a Ki-67 index of >20%
- Synaptophysin negative tumors with a Ki-67 index of >20%
- Synaptophysin positive tumors with a Ki-67 index of >20%
- Well-differentiated tumors with a Ki-67 index of >20%
- Which of the following is true of the clear cell variant of PanNET?
- Commonly seen in Von Hippel-Lindau syndrome
- Infiltrating borders are characteristic
- Mucin positive tumor
- Single prominent nucleolus is typical
- Synaptophysin negative tumor
References
- Fryer E, Serra S, Chetty R. Lipid-rich ("clear cell") neuroendocrine tumors of the pancreas in MEN I patients.Endocr Pathol. 2012;23(4):243-246.
- Hackeng WM, Hruban RH, Offerhaus GJ, Brosens LA. Surgical and molecular pathology of pancreatic neoplasms. Diagn Pathol. 2016;11(1):47.
- Hoang MP, Hruban RH, Albores-Saavedra J. Clear cell endocrine pancreatic tumor mimicking renal cell carcinoma: a distinctive neoplasm of von Hippel-Lindau disease. Am J Surg Pathol. 2001;25(5):602-609.
- Kloppel G, Couvelard A, Hruban RH et al. Neoplasms of neuroendocrine pancreas. In Lloyd RV, Osamura RY, Kloppel G and Rosai J (eds). WHO classification of Tumours of Endocrine Organs. Lyon, FR: IARC press; 2017:210-221.
Author
Vijaya B. Reddy, MD
Surgical Pathology Committee
Rush University Medical Center
Chicago, IL
Answer Key
- Labeling for nuclear β-catenin (c)
- Well-differentiated tumors with a Ki-67 index of >20% (e)
- Commonly seen in Von Hippel-Lindau syndrome (a)