- Home
- Member Resources
- Pathology Case Challenge
- Posterior Mediastinal Mass
Clinical Summary
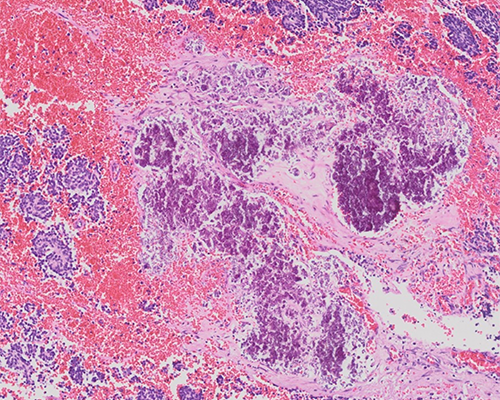
A 9-month-old infant develops respiratory distress and is found to have a left posterior mediastinal mass and pleural effusion. On excision, the specimen consists of a smoothly lobulated, well-circumscribed mass measuring 5.0 x 3.5 x 1.7 cm. Immunohistochemistry reveals the tumor to be positive for NSE and chromogranin and negative for CD45 and desmin. Scattered S-100 positive stroma cells are also noted.
Master List of Diagnoses
- Desmoplastic small round cell tumor
- Ewing sarcoma
- Lymphoblastic lymphoma
- Neuroblastoma
- Rhabdomyosarcoma
Archive Case and Diagnosis
This case first appeared as Performance Improvement Program in Surgical Pathology (PIP) 2018, Case 14, and is a poorly-differentiated neuroblastoma found in the posterior mediastinum.
Criteria for Diagnosis and Comments
The histologic sections reveal small blue cells arranged in nests and sheets separated by thin fibrovascular septa. Fine pink fibrillar material consistent with neuropil is readily identified. Pseudorosettes (Homer Wright rosettes) are also noted. While rare neuroblasts show an early attempt toward ganglion cell differentiation, this is not prominent and comprises less than 5% of the population. Calcification and scattered lymphoid aggregates are also noted. The mitotic karryorhectic index (MKI) is intermediate (100 to 200 per 5,000 cells).
The histologic features are those of poorly-differentiated neuroblastoma. The presence of neuropil helps exclude other small blue cell tumors. Clinical and radiographic features can also strongly suggest a neuroblastoma (elevated urinary catecholamines, uptake of metaiodobenzylguanidine or MIBG by scintigraphy).
Neuroblastoma is the most common extracranial solid tumor in childhood. The overall incidence is about 1 case per 100,000 children in the United States. Approximately 90% of tumors arise in children under the age of 10 years. The median age at diagnosis is 18 months. Neuroblastoma is slightly more common in males than females.
Neuroblastoma arises from neural crest cells present in the developing sympathetic nervous system. Tumors arise in the migratory pathway of the neural crest including the adrenal medulla and along the sympathetic chain. Neuroblastoma has some unique clinical and biological features that distinguish it from other tumors. These include its early age of onset, the high frequency of metastasis at the time of presentation, and its tendency for spontaneous regression in young children. Prognosis is related to the age of the patient, histologic subtype, stage, tumor location, and genetic and laboratory factors. Of note, familial cases have been identified in 1% to 2% of cases. The two genes associated with this are ALK (anaplastic lymphoma kinase) and PHOX2B, a transcription factor involved in neural crest development. ALK has been found to be mutated in both hereditary and sporadic tumors whereas mutated PHOX2B has only rarely been noted in non-hereditary tumors.
Neuroblastic tumors are biologically and histologically diverse, ranging from tumors that regress, to tumors that show an age-matched maturation, to tumors which are highly aggressive. Dr. Shimada proposed a classification for neuroblastoma incorporating the age of the child and the mitotic karyorrhectic index (MKI) of the tumor. This scheme was modified in 2001 by the International Neuroblastoma Pathology Committee into four categories of neuroblastoma. Each tumor is evaluated for four features: the presence or absence of Schwannian stroma, the degree of differentiation of the neuroblasts, the MKI, and the uniformity of the tumor. Examining stroma and neuroblastic differentiation yields four main categories: neuroblastoma, a tumor which is lacking in Schwannian stroma (stroma-poor); ganglioneuroblastoma-intermixed, a tumor which is Schwannian stroma-rich and contains less than 50% neuroblastic differentiation; a ganglioneuroma which is Schwannian stroma-dominant; and nodular ganglioneuroblastoma (GNBn) (composite stroma-rich/stroma-dominant and stroma-poor). In GNBn the stroma-rich component and the neuroblastic component are distinctly separate areas.
From these groups, favorable and unfavorable subcategories are created by evaluating low, intermediate, or high MKI; neuroblastic differentiation; MYCN status; and patient age. MKI assessments are made by counting mitoses and karyorrhectic cells per 5,000 cells. MKI is assessed by counting multiple fields and averaging the results. The MKI is based on the overall average of the tumor not on the area with the highest number of mitoses or karyorrhectic cells. A low MKI corresponds to less than 2% or less than100 MK cells per 5,000 cells, intermediate to 2% to 4% or 100 to 200 MK cells per 5,000 cells, and high 4% or greater than 200 MK cells per 5,000 cells.
Neuroblastic differentiation is categorized as undifferentiated (no evidence of stroma or of ganglion cell differentiation), poorly-differentiated with neuropil present but < 5% differentiating neuroblasts and differentiating neuroblastoma with greater than 5% differentiating neuroblasts. Highly prognostic groups can be identified when these features are associated with the patient’s age. Therefore, poorly-differentiated tumors and differentiating tumors with low or intermediate MKI in children less than 1.5 years of age and differentiating tumors with a low MKI in children 1.5 to 5 years of age are associated with an excellent prognosis (favorable histology). A poor prognosis (unfavorable histology) is associated with undifferentiated tumors at any age, high MKI at any age, poorly-differentiated tumors and differentiating tumors with an intermediate MKI in patients greater than 1.5 years of age, and all neuroblastomas in patients greater than 5 years of age. Evaluating the neuroblastic differentiation and MKI status of the neuroblastic or stroma poor component of a nodular GNBn also allows further sub classification of GNBn to favorable and unfavorable subtypes.
In 2007, Tornoczky et al. identified a subset of neuroblastoma which they called “large cell neuroblastoma”. These tumors were poorly-differentiated or undifferentiated neuroblastomas found to contain neuroblasts with large prominent nucleoli, a feature which is often associated with differentiation and maturation to a ganglion cell. In these cases, however, these cells did not show the necessary cytoplasmic maturation that accompanies the nuclear changes.
MYCN belongs to the Myc family of proto-oncogenes, which are important in development and in the regulation of cell proliferation and apoptosis. Amplification of MYCN has been identified in 20% to 25% of cases of neuroblastoma. Amplified MYCN results in increased expression of MDM2 and TP53, both of which sensitize the tumor cells to apoptosis. Incorporating MYCN status with morphology creates four subsets: favorable histology (FH)-unamplified, FH-amplified, unfavorable histology (UF)-unamplified, UF-amplified. Most FH tumors are unamplified and have an excellent prognosis. These tumors also tend to be stage 1 or stage 4s, a special category defined as children less than 1 year of age with metastases limited to the skin, liver, or bone marrow. The rare patients with FH-amplified have a worse prognosis with higher stage and more aggressive behavior. However, these tumors do respond to therapy. Patients with UH-amplified have the highest stage of disease and the worst prognosis.
MYCN amplification is usually associated with segmental chromosomal loss of the distal short arm of chromosome 1. Gain of 17q has been identified in over half of cases of neuroblastoma and is also associated with MYCN amplification and poor prognosis. Loss of 11q has been found in one-third of high-risk neuroblastoma and is inversely correlated with MYCN amplification. In infants, hyperdiploid tumors seem to respond better to therapy and are considered a favorable prognostic category.
The separation of undifferentiated neuroblastoma from other small blue cell tumors requires immunohistochemistry and/or molecular or genetic testing. Until recently, there has not been a specific marker for neuroblastoma. Chromogranin and synaptophysin can be useful but can show variable staining. Schwannian stroma in the fibrovascular septa may be positive for S100. INSM-1 is a relatively new marker for neuroendocrine tumors and can be positive in neuroblastoma. Additional neuronal markers which are useful include protein gene product 9.5 (PGP9.5), neuron specific enolase, NB84, CD56, CD57, and tyrosine hydroxylase. However, PGP9.5 and INSM-1 are not specific for neuroblastoma and may stain other tumors with neuroectodermal differentiation, including Ewing sarcoma. Recently PHOX2B, derived from the PHOXB2 gene, which is highly specific for autonomic nervous system development, has been found strongly and uniformly expressed in neuroblastoma, and preliminary studies show it being relatively specific. Recent studies have also confirmed its usefulness in identifying bone marrow metastases and post therapy residual disease, on par with CD57.The benefit to PHOX2B is that it is a nuclear stain, which can make it easier to interpret.
The differential diagnosis of neuroblastoma includes other small blue cell tumors of childhood, especially Ewing sarcoma, lymphoma/leukemia, rhabdomyosarcoma, and desmoplastic small round cell tumor (DSRCT). There is overlap with some immunohistochemical markers and not all of them are readily available in all laboratories. Molecular analysis is considered the gold standard and is therefore very important. The following markers are negative in neuroblastoma but are positive in tumors in the differential diagnosis:
Rhabdomyosarcoma is favored when myogenin, desmin, and Myo D1 are expressed.
Ewing sarcoma is favored when NKX2.2 is expressed and there is and membranous staining of CD99. most Ewing sarcoma show an EWS-FLI-1 or EWS-ERG fusion.
DSRCT is supported when there is co-expression of cytokeratin markers, vimentin, and desmin. DRSCT also expresses nuclear WT-1 and is negative for chromogranin and synaptophysin. The identification of an EWS-WT1 t(11;22)(q13;q12) translocation is diagnostic of DSRCT.
Most types of lymphoma can be excluded by a negative CD45. TdT is useful to rule out lymphoblastic lymphoma, which usually expresses TdT but can be negative for CD45.
Supplementary Questions
- A 5-year-old boy presents with abdominal swelling. Biopsy reveals a small blue cell tumor that is positive for NKX2.2 and CD99 in a membranous pattern. Desmin and cytokeratin are negative.
Which of the following is the most likely diagnosis?
- Desmoplastic small round cell tumor
- Ewing sarcoma
- Lymphoblastic lymphoma
- Neuroblastoma
- Wilms tumor
- Which of the following are the two best antibodies to use to detect bone marrow metastases and minimal residual disease of neuroblastoma?
- CD56 and CD99
- NB84 and S100
- PHOX2B and NB84
- S100 and NSE
- TDT and CD45
- Which of the following features is associated with unfavorable histology in neuroblastoma in a 1-year-old child?
- Differentiating neuroblastoma, high mitotic karyorrhectic index (MKI)
- Ganglioneuroblastoma, intermixed
- Hyperdiploid neuroblastoma
- Nodular ganglioneuroblastoma where the nodule is low MKI
- Poorly-differentiated neuroblastoma, intermediate MKI, MYCN non-amplified
References
- Hata JL, Correa H, Krishnan C, et al. Diagnostic utility of PHOX2B in primary and treated neuroblastoma and in neuroblastoma metastatic to the bone marrow. Arch Pathol Lab Med. 2015;139:543-546.
- Matthay KK, Maris JM, Schleiermacher G, et al. Neuroblastoma. Nat Rev Dis Primers 2016;2:16078.
- Peuchmaur M, d'Amore ES, Joshi VV, et al. Revision of the International Neuroblastoma Pathology Classification: confirmation of favorable and unfavorable prognostic subsets in ganglioneuroblastoma, nodular. Cancer. 2003;98:2274-2281.
- Picarsic J, Reyes-Mugica M. Phenotype and immunophenotype of the most common pediatric tumors. Appl Immunohistochem Mol Morphol. 2015;23:313-326.
- Shimada H, Umehara S, Monobe Y, et al. International neuroblastoma pathology classification for prognostic evaluation of patients with peripheral neuroblastic tumors: a report from the Children's Cancer Group. Cancer. 2001;92:2451-2461.
- Suganuma R, Wang LL, Sano H, et al. Peripheral neuroblastic tumors with genotype-phenotype discordance: a report from the Children's Oncology Group and the International Neuroblastoma Pathology Committee. Pediatr Blood Cancer. 2013;60:363-370.
- Teshiba R, Kawano S, Wang LL, et al. Age-dependent prognostic effect by Mitosis-Karyorrhexis Index in neuroblastoma: a report from the Children's Oncology Group. Pediatr Dev Pathol. 2014;17:441-449.
- Thway K, Noujaim J, Zaidi S, et al. Desmoplastic small round cell tumor: pathology, genetics, and potential therapeutic strategies. Int J Surg Pathol. 2016;24:672-684.
- Tornóczky T, Kalman E, Kajtar PG, et al. Large cell neuroblastoma: a distinct phenotype of neuroblastoma with aggressive clinical behavior. Cancer. 2004;100:390-397.
- Warren M, Shimada H. Importance of Phox2B immunohistochemical stain for detecting metastatic neuroblastoma cells in bone marrow specimens. Pediatr Dev Pathol. 2016;19:254-255.
Authors
Deborah Ann Belchis, MD
Surgical Pathology Committee
Johns Hopkins Bayview Med Center
Baltimore, MD
Answer Key
- Ewing sarcoma (b)
- PHOX2B and NB84 (c )
- Differentiating neuroblastoma, high mitotic karyorrhectic index (MKI) (a)