- Home
- Member Resources
- Pathology Case Challenge
- Adrenal Gland
Clinical Summary
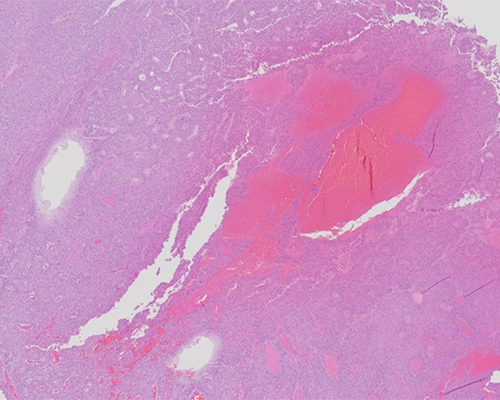
A 56-year-old woman presents with abdominal pain, bloating, and early satiety of several months’ duration. She also reports cramps, weight gain, and palpitations over this same period. She is found to have elevated DHEA-S and testosterone. Imaging shows a large right adrenal mass, which is resected. Gross examination reveals a 12 cm yellow-tan mass with areas of necrosis. The mass grossly invades the adjacent adrenal gland.
Master List of Diagnoses
- Adrenal cortical adenoma
- Adrenal cortical carcinoma
- Metastatic mammary carcinoma
- Metastatic melanoma
- Pheochromocytoma
Archive Case and Diagnosis
This case first appeared as Performance Improvement Program in Surgical Pathology (PIP) 2018, Case 23, and is an adrenal cortical carcinoma of the adrenal gland.
Criteria for Diagnosis and Comments
Histologically, the tumor is composed of sheets of cells with nuclear pleomorphism and areas of oncocytic appearance. The cells have prominent nucleoli and areas of necrosis and mitoses are present. By immunohistochemistry, the tumor cells are positive for inhibin and negative for chromogranin and S100. These findings are consistent with adrenal cortical carcinoma (ACC).
ACC is composed of epithelial cells originating from the adrenal cortex. Oftentimes, ACCs have thick capsules, broad fibrous bands, tumor cells arranged in disordered nests and trabeculae, and necrosis. The current World Health Organization (WHO) endocrine edition distinguishes adult ACC from adrenal adenoma based on the Weiss criteria. The Weiss criteria define malignancy based on the presence of three or more of the following features: high-grade nuclei, high mitotic rate (more than 5 mitoses per 50 high-power fields (HPF)), atypical mitotic figures, clear cells comprising less than 25% of cells, diffuse architecture, tumor necrosis, venous invasion, sinusoidal invasion, and capsular invasion. Special stains such as reticulin can be used to highlight disruption of nested architecture. Immunohistochemical stains, including laminin and collagen type IV, can be employed to highlight interruption of the basal lamina and document the diffuse architecture seen on hematoxylin and eosin stained slides. The Weiss criteria are not reliable indicators of malignancy in pediatric patients. In this population, features of malignancy include large tumor size (weight greater than 400 grams and tumor size greater than 10.5 cm), vena cava invasion, confluent necrosis, higher mitotic rate (more than 15 mitoses per HPF), atypical mitoses, and invasion of adjacent soft tissue.
Of primary adrenal tumors, ACC is less common than cortical adenoma and pheochromocytoma. Clinically, ACC presents more commonly in women and has a bimodal distribution, occurring in the first and fifth decades of life. Half of the patients with ACC exhibit symptoms and signs of excess steroid hormone production, while the remaining are asymptomatic at the time of diagnosis or experience mechanical effects related to tumor growth. Amongst functioning tumors, cortisol-producing ACC is more common than aldosterone or sex hormone secreting tumors.
Advanced imaging techniques often reveal adrenal tumors in asymptomatic patients. Incidentally found adrenal masses can be further classified as benign or malignant and primary adrenal versus metastatic using radiologic-cytologic or pathologic correlation. Although vascular and capsular invasion cannot be assessed, cytologic features on fine needle aspiration (FNA) such as hypercellularity, necrosis, and cellular dyscohesion, in conjunction with radiologic features of tumor size (greater than 4.0 cm), heterogeneity, and enhancement, favor ACC over other adrenal neoplasms. Radiologic and FNA cytologic studies are also useful in staging patients with possible metastatic lesions.
Grossly, most ACCs weigh greater than 100 grams and appear either encapsulated or are adherent to/infiltrating into adjacent structures. The fleshy pink-brown cut surface exhibits a lobulated parenchyma separated by fibrous septa. Foci of hemorrhage and necrosis are commonly seen and a subset exhibit cystic change or vascular invasion.
It is important to distinguish the histological variants of conventional ACC as they share similar features to secondary tumors that not infrequently involve the adrenal gland. The most common variant, oncocytic ACC, is characterized by cells with abundant eosinophilic cytoplasm. This variant should be differentiated from oncocytic adenoma which may exhibit atypical features that are components of the Weiss criteria including high nuclear grade, less than 25% clear cells, and diffuse architecture. Thus, an alternative scoring system (Li-Weiss-Bisceglia system) is recommended for oncocytic tumors: high mitotic rate (more than 5 mitoses per 50 HPF), atypical mitotic figures, and venous invasion favor oncocytic ACC over oncocytic adenoma. Additionally, Ki-67 proliferation index greater than 5% by immunohistochemistry can help differentiate ACC from adrenal adenoma. The myxoid and sarcomatoid variants of ACC are rare but pose potential diagnostic pitfalls. Myxoid ACC demonstrates abundant stromal extracellular mucin and tends to be underdiagnosed using Weiss criteria. Sarcomatoid ACC displays biphasic morphology and may lack regions of conventional ACC and easily mistaken for retroperitoneal sarcoma involving the adrenal gland. Adrenal cortical tumors are positive for inhibin, calretinin, and Melan-A, though these stains do not distinguish between benign and malignant tumors.
ACCs harbor a variety of molecular alterations, the most prevalent of which are IGF2 overexpression and TP53 mutations. TP53 mutations are the dominant mutations in pediatric cases and herald a poor prognosis. IGF2 gene overexpression and rearrangement at the 11p15.5 locus is present in the majority of sporadic ACC cases and paternal disomy of the IGF2 gene is seen in ACC occurring in patients with Beckwith-Wiedemann syndrome. TP53 mutations in ACC may be germline (seen in Li-Fraumeni syndrome) or sporadic and are rarely seen in adrenal adenomas. Molecular mechanisms driving disease progression include copy number alterations, whole genome doubling, and telomere and telomerase activation. Currently, the lack of targetable mutations and poor response to chemotherapy due to inherent forms of drug resistance make complete surgical excision the mainstay of treatment. Unfortunately, 15% of ACCs are locally invasive and 67% have metastasized at the time of diagnosis, further compounding the poor prognosis of this tumor. Median survival is 4 to 30 months from diagnosis and mortality from ACC is 67% to 94%.
The staging system for ACC recommended by the WHO is based on the European Network for the Study of Adrenal Tumors, which classifies ACC based on size and extent of the tumor. Organ confined stages, stages I and II, include tumors smaller than 50 mm and larger than 50 mm in diameter, respectively. Stage III tumors involve surrounding tissue, regional lymph nodes, or regional veins. Tumors with distant metastases are classified as stage IV.
The differential diagnosis of ACC includes other primary adrenal cortical tumors, including adrenal cortical adenoma and pheochromocytoma, as well as metastatic tumors, including breast carcinoma and melanoma.
Like ACC, adrenal cortical adenomas may be functioning or nonfunctioning and are composed of cells with abundant intracytoplasmic lipid. The Weiss criteria are used to distinguish adrenal cortical adenomas from carcinomas. While ACC and adrenal cortical adenomas share immunohistochemical expression patterns, they may be characterized by different molecular profiles. For example, cortisol secreting adrenal cortical adenomas may harbor PRKAR1A mutations which are seen in association with Carney complex.
Pheochromocytomas are intra-adrenal paragangliomas and share some histologic and immunophenotypic features with ACC. Pheochromocytomas are composed of cells with granular cytoplasm, nuclear pseudoinclusions, and nuclear pleomorphism. They have a vascular stroma and sometimes contain melanin and other pigments. To distinguish ACC from pheochromocytoma, laboratory data (hormone levels) and clinical signs (hypertension) can be helpful. Synaptophysin and neurofilament protein typically present in pheochromocytoma have also been reported in ACC, especially the myxoid type. Expression of calretinin, inhibin, SF1 and/or Melan-A support a diagnosis of ACC, as these stains are negative in pheochromocytomas. Chromogranin-A reactivity favors a diagnosis of pheochromocytoma over ACC. Further confirmation may be sought ultrastructurally as ACC distinctly features abundant smooth endoplasmic reticulum and mitochondria, as well as prominent tubular cristae.
Differentiating metastatic melanoma from ACC, particularly the sarcomatoid variant, can be difficult on histology. ACC and metastatic melanoma both express Melan-A, creating potential confusion. However, expression of S100 and HMB-45 and negative staining with SF-1 and inhibin favor metastatic melanoma over ACC.
Poorly differentiated metastatic mammary carcinoma can be difficult to distinguish from ACC. Negative staining with SF-1, inhibin, and Melan-A, and expression of GATA-3 with or without ER/PR expression, depending on the hormone receptor status of the primary tumor, favor metastatic mammary carcinoma.
Supplementary Questions
- Which of the following immunohistochemical stains, if positive, would favor a diagnosis of pheochromocytoma over adrenal cortical carcinoma (ACC)?
- Calretinin
- Chromogranin-A
- Inhibin
- Melan-A
- SF-1
- Which of the following statements is the most accurate?
- ACCs arise in patients with Beckwith-Wiedemann syndrome.
- ACCs arise in patients with Cushing syndrome.
- ACCs arise in patients with multiple endocrine neoplasia type 2 (MEN2).
- ACCs arise in patients with neurofibromatosis type 1 (NF1).
- ACCs arise in patients with Von Hippel-Lindau (VHL) syndrome.
- Which of the following statements is true?
- ACC is the most common primary tumor of the adrenal gland.
- ACC occurs more commonly in males.
- ACC presents in 50% of cases with signs and symptoms of excess steroid hormone production.
- ACCs are highly chemosensitive.
- ACCs are typically smaller on radiologic scans than adrenal adenomas.
References
- Duregon E, Volante M, Bollito E. Pitfalls in the diagnosis of adrenocortical tumors: a lesson from 300 consultation cases. Hum Pathol. 2015;46(12):1799-1807.
- Lam AK. Update on Adrenal Tumours in 2017 World Health Organization (WHO) of Endocrine Tumours. Endocr Pathol. 2017:27(3):213-227.
- Lattin GE Jr, Sturgill ED, Tujo CA. From the radiologic pathology archives: Adrenal tumors and tumor-like conditions in the adult: radiologic-pathologic correlation. Radiographics. 2014;34(3):805-829.
- Lloyd RV. Adrenal cortical tumors, pheochromocytomas and paragangliomas. Modern Pathol. 2011:24 Suppl 2:S58-S65.
- McNicol AM. Lesions of the Adrenal Cortex. Arch Pathol Lab Med. 2008;132(8):1263-1271.
- Ren R, Guo M, Sneige N, Moran CA, Gong Y. Fine-needle aspiration of adrenal cortical carcinoma: cytologic spectrum and diagnostic challenges. Am J Clin Pathol. 2006;126(3):389-398.
Authors
Rachel Jug, MB BCh BAO
Pathology Resident
Duke Health
Durham, NC
Xiaoyin “Sara” Jiang, MD
Surgical Pathology Committee
Duke Health
Durham, NC
Answer Key
- Chromogranin-A (b)
- ACCs arise in patients with Beckwith-Wiedemann syndrome. (a)
- ACC presents in 50% of cases with signs and symptoms of excess steroid hormone production. (c)