Clinical Summary
A 6-month-old baby boy presents to an outside hospital with cardiomyopathy of uncertain etiology. The baby is transferred to a tertiary care center where cardiac transplantation is performed. Representative sections of the explanted heart are submitted. PAS stains with and without diastase show no increase in intracytoplasmic glycogen or increase of PAS-positive diastase resistant material. A modified Gomori trichrome stain shows granular staining of the cytoplasm. Diagnostic electron microscopy reveals increased numbers of an abnormal organelle.
Master List of Diagnoses
- Arrhythmogenic cardiomyopathy
- Glycogen storage disorder
- Mitochondrial cardiomyopathy
- Mucopolysaccharidoses
- Myocarditis
Archive Case and Diagnosis
This case first appeared as Performance Improvement Program in Surgical Pathology (PIP) 2019, Case 03, and is mitochondrial cardiomyopathy (heart).
Criteria for Diagnosis and Comments
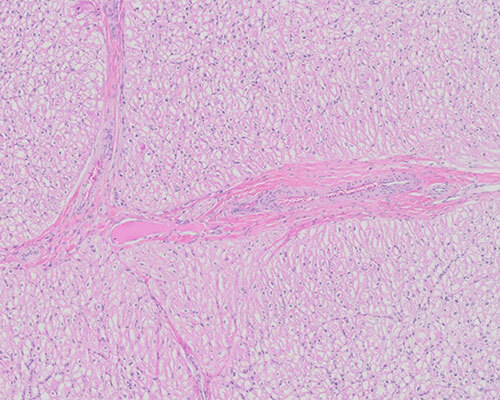
H&E sections of the heart show myocytes with reactive nuclear changes characterized by mild pleomorphism, hyperchromasia, and enlargement, and prominent vacuolar cytoplasmic change. Intracytoplasmic membrane-bound vacuoles are absent. There is no evidence of myocyte disarray or inflammation.
PAS stains with and without diastase show no increase in intracytoplasmic glycogen or diastase-resistant material. A modified Gomori trichrome stain shows granular staining of the cytoplasm correlating with abnormal mitochondria identified by electron microscopy. A toluidine blue stain highlights distended and displaced myofilaments within the cytoplasm of the myocytes.
Electron microscopy reveals increased numbers of mitochondria within the cytoplasm. Many of the mitochondria are dysmorphic, consisting of circular, concentrically (onion-like) arranged cristae and some arranged in parallel arrays. No abnormal metabolic product is identified. The overall changes are diagnostic of mitochondrial cardiomyopathy. The etiology of the mitochondrial cardiomyopathy cannot be determined from the morphologic features alone.
Cardiomyopathy (CMP) is a rare disorder in children with an estimated 1.3 cases annually per 100,000 in those less than 10 years of age. It is associated with high morbidity and mortality. Recent studies have found up to 26% of pediatric cardiomyopathy cases to be due to inborn errors of metabolism, a subset of which fall into the category of mitochondrial disorders. Mitochondrial diseases are a clinically, genetically, and biochemically heterogeneous group of disorders. Mitochondria are important providers of energy as they generate adenosine triphosphate via the electron transport chain (ETC) and oxidative phosphorylation system. Disorders of mitochondria, therefore, preferentially affect organ systems with high energy requirements such as the heart, brain, and neuromuscular systems. Mitochondrial disorders may be primary or secondary. Primary disorders are due to mutations in mitochondrial DNA or nuclear DNA. Most mitochondrial myopathies are non-Mendelian, since mitochondrial DNA is passed maternally with the cytoplasm and not in the nucleus of dividing cells. Secondary mutations are the result of an insult affecting mitochondrial function either directly or indirectly or by damaging the mitochondrial genome. Inadequate levels of mitochondrial DNA and its mutations can cause defects in the synthesis of key subunits of ETC complexes.
The clinical presentation of mitochondrial myopathy is highly variable. Muscle fatigue and weakness, which can manifest in infants as failure to thrive, are frequent symptoms and are easily overlooked. There is great clinical variability in presentation, even for the same mutations, making diagnosis difficult. The prevalence of inherited mitochondrial disease has been estimated at 1 in 5,000. Cardiomyopathy develops in 20% to 40% of children with mitochondrial disease.
The morphologic features of the heart in mitochondrial CMP are not specific, leading to a broad differential diagnosis including storage, metabolic, genetic, and environmental disorders. Hypertrophic CMP is the most common manifestation of mitochondrial CMP; however, dilated CMP, restrictive CMP, histiocytoid CMP, or left ventricular hypertrabeculation/noncompaction (LVHT) CMP may also occur. LVHT is characterized by prominent ventricular trabeculations or channels that extend into the myocardium, creating deep recesses within the myocardium.
Mitochondrial diseases may be hereditary or sporadic. Mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes (MELAS), myoclonic epilepsy with ragged red fibers (MERRF), chronic progressive external ophthalmoplegia, Leigh syndrome, X-linked cardiomyopathy, mitochondrial myopathy, cyclic neutropenia (Barth syndrome), and neuropathy, ataxia, and retinitis pigmentosa (NARP) are examples of syndromic mitochondrial disorders causing hypertrophic CMP. Skeletal muscle biopsy is considered the gold standard for the diagnosis of mitochondrial disorders. Ragged red fibers are considered the histologic hallmark of mitochondrial dysfunction, but these are not typically seen in children.
Microscopic features of mitochondrial myopathy vary. Fusiform enlargement of affected myocytes around the perinuclear region with cytoplasmic clearing and replacement of cross striae by fine granules may suggest a mitochondrial abnormality. Excessively granular eosinophilic cytoplasm of hepatocytes, cardiac myocytes, skeletal muscle, and proximal renal tubules also may indicate mitochondrial hyperplasia.
Laboratory workup includes analysis for lactate, pyruvate, phosphocreatine, amino acid abnormalities, organic acids, creatinine, and quantification of coenzyme Q. The diagnosis may be made by special stains including cytochrome oxidase (oxidase-negative myocytes), mitochondrial antigens that can demonstrate mitochondrial hyperplasia, and staining for succinate dehydrogenase. Postmortem analysis of urine and vitreous fluid, frozen tissue, and a fibroblast culture provide a comprehensive study. Genomic and/or mitochondrial sequencing is increasingly being used for diagnosis. Ultrastructural analysis will show paracrystalline inclusions, enlarged mitochondria, and concentric onion-like cristae.
The differential diagnosis of mitochondrial myopathy includes glycogen storage diseases, mucopolysaccharidoses, myocarditis, and arrhythmogenic cardiomyopathy.
Glycogen storage diseases (GSD) are inherited disorders due to a defect in enzymes that are involved in glycogen metabolism. The two types most commonly affecting the heart are type IIa and IIb GSD. Infants with type IIa (Pompe disease), which is due to deficiency of alpha-1,4, alpha 1-6 glucosidase (acid maltase), manifest disease in infantile or late forms depending on the severity of the enzymatic defect. There is left ventricular hypertrophy. Grossly, the heart can weigh 3-10 times the normal size and show a globular configuration. Microscopically the myocytes are markedly distended with vacuolated and lacey cytoplasm due to the accumulation of PAS-positive glycogen. Ultrastructural examination shows the glycogen to be at least partially membrane bound. Type IIb (Danon disease) is an X-linked disease caused by a deficiency of lysosome-associated membrane protein-2 (LAMP-2). Affected children present with hypertrophic CMP, muscle weakness, and mental retardation (70%). Microscopic examination reveals a vacuolar myopathy. The myocytes contain PAS- and acid phosphatase-positive inclusions. Ultrastructural examination shows intracytoplasmic membrane-bound vacuoles containing glycogen.
Mucopolysaccharidoses are lysosomal storage disorders that commonly affect the heart manifested by valvular insufficiency. Irregular nodular deposits are present along the valves. There is intimal plaque formation in the aorta and other systemic vessels. Microscopy reveals vacuolated cells containing acid mucopolysaccharides and glycolipids. Ultrastructurally, membrane-bound vacuoles contain concentric and parallel lamellae.
Myocarditis, or inflammatory CMP, is an inflammatory disease of the myocardium and can be infectious, autoimmune, allergic, or drug induced and is classified under the acquired forms of cardiomyopathy. It is diagnosed by endomyocardial biopsy, which shows inflammation with or without myocyte necrosis. It can be further subdivided according to the type of inflammatory cell present: neutrophils, eosinophils, or lymphocytes. It can resolve or progress to dilated CMP. The most common cause of inflammatory CMP in the United States is a lymphocytic myocarditis due to a virus, typically Coxsackie viruses A and B or another enterovirus.
Arrhythmogenic cardiomyopathy is an autosomal dominant disorder that manifests itself with right-sided heart failure and rhythm abnormalities. Morphologically, the right ventricular wall is thinned with fatty infiltration and mild fibrosis. Our understanding of the underlying genetic causes of arrhythmogenic CMP is evolving with next generation sequencing and other molecular and genetic analyses. Mutations in genes involved in desmosomal junctional proteins are well recognized to be associated with the development of arrhythmogenic CMP. Recently mutations involving nondesmosomal genes, such as mutations in transforming growth factor beta, have also been identified in families with a history of arrhythmogenic CMP.
Supplementary Questions
- A mother brings her child to the pediatrician stating that the child is easily tired. The child exhibits developmental delay. Cardiac examination reveals a hypertrophic cardiomyopathy. A cardiac biopsy shows vacuolated myocytes that are PAS positive. Acid maltase activity levels were normal. Which of the following is the best diagnosis?
- A mitochondrial disorder
- Arrhythmogenic cardiomyopathy
- Type IIa glycogen storage diseases (Pompe disease)
- Type IIb glycogen storage disease (Danon disease)
- Viral myocarditis
- Which of the following most accurately describes mitochondrial disorders?
- Morphologically, the right ventricle is thinned with fatty infiltration and fibrosis.
- Ragged red fibers are not associated with mitochondrial cardiomyopathy.
- Stains for cytochrome oxidase and mitochondrial antigens are useful in making the diagnosis.
- They usually present with dilated cardiomyopathy.
- They are easily recognized disorders due to the specific symptomatology.
- Which of the following is the most frequent pattern of inherited mitochondrial myopathies?
- Autosomal dominant
- Autosomal recessive
- Non-Mendelian
- X-linked dominant
- X-linked recessive
References
- Byers SL, Ficicioglu C. Infant with cardiomyopathy: when to suspect inborn errors of metabolism? World J Cardiol. 2014;6(11):1149-1155.
- Dewulf JP, Barrea C, Vincent M-F, et al. Evidence of a wide spectrum of cardiac involvement due to ACAD9 mutations: report on nine patients. Molecular Genetics and Metabolism. 2016; 118(3):185-189.
- Karnouch J, Protonotarios A, Syrris P. Genetic basis of arrhythmogenic cardiomyopathy. Curr Opin Cardiol. 2018;33(3):276-281.
- Meyers DE, Basha HI, Koenig MK. Mitochondrial cardiomyopathy: pathophysiology, diagnosis, management. Tex Heart Inst J. 2013;40(4):385-394.
- Sweeney RT, Davis GJ, Noonan JA. Cardiomyopathy of unknown etiology: Barth Syndrome unrecognized. Congenit Heart Dis. 2008;3:443-448.
- Towbin JA. Inherited cardiomyopathies. Circ J. 2014;78(10):2347-2356.
- Wallis G, Fricker FJ. Neonatal cardiomyopathy. NeoReviews. 2012;13:e711-e721.
Author
Deborah A. Belchis, MD
Surgical Pathology Committee
Johns Hopkins Medical Institution
Baltimore, Maryland
Answer Key
- Type IIb glycogen storage disease (Danon disease) (d)
- Stains for cytochrome oxidase and mitochondrial antigens are useful in making the diagnosis. (c)
- Non-Mendelian (c)