Clinical Summary
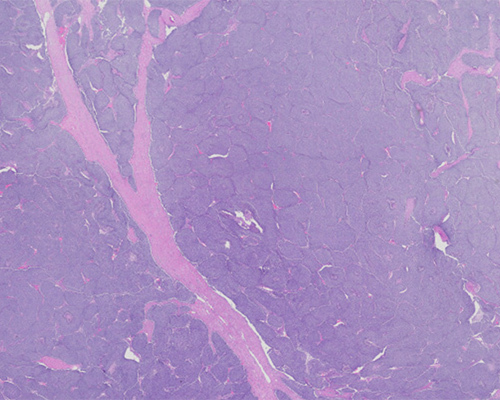
A 4-year-old girl is found to have an abdominal mass. Computed tomography scan confirms the presence of a solid and focally cystic left renal mass. The resection specimen shows a well-circumscribed subcapsular intrarenal mass that involves the upper pole of the left kidney, measures 18 cm in greatest dimension, and exhibits a variegated pink-tan, soft, and focally friable cut surface. A central hemorrhagic and multicystic area measures 6 cm in greatest dimension.
Master List of Diagnoses
- Clear cell sarcoma of kidney
- Congenital mesoblastic nephroma
- Intrarenal neuroblastoma
- Nephroblastoma (Wilms tumor), blastema-predominant
- Rhabdoid tumor of kidney
Archive Case and Diagnosis
This case first appeared as Performance Improvement Program in Surgical Pathology (PIP) 2019, Case 23, and is Nephroblastoma (Wilms tumor), blastema-predominant.
Criteria for Diagnosis and Comments
Upon histologic evaluation, the renal tumor is characterized by a predominant proliferation of blastema arranged in serpentine and nested patterns. Most of the blastemal component is characterized by a proliferation of “small blue cells” with high nuclear-to-cytoplasmic ratios, coarse chromatin, and brisk mitotic activity. The epithelial component consists mainly of a few tubules lined by primitive cuboidal to columnar cells, and the stromal component is variably edematous. Areas of tumor necrosis are noted. A diagnosis of nephroblastoma or Wilms tumor (WT), blastema-predominant, is established.
In the United States, nephroblastoma (Wilms tumor) is the most common primary malignant renal tumor in the pediatric age group. The peak incidence of this neoplasm is between 2 and 5 years of age, with approximately 95% of cases occurring before the age of 10. In close to 10% of cases, WT arises in association with three syndromes: WAGR (Wilms tumor, Aniridia, Genital anomalies, and intellectual disability [formerly referred to as mental Retardation]) syndrome, Denys–Drash syndrome (gonadal dysgenesis and early onset nephropathy), and Beckwith–Wiedemann syndrome (organomegaly, macroglossia, hemihypertrophy, omphalocele, and adrenal cytomegaly). Although WT usually presents as a painless abdominal mass, in close to a third of cases, the clinical presentation may include anorexia, pain, and vomiting. In some patients (especially young children), renal masses can be detected by palpation.
Grossly, WT usually presents as a single well-circumscribed mass with a pseudocapsule. Multifocal and bilateral tumors are seen in less than 10% of patients. The cut surface is lobulated, pale gray or pink, soft, and friable. Common findings include hemorrhage, necrosis, calcification, and cystic change. Extension into the renal pelvis and ureter can be seen, and invasion of the renal vein is often identified.
Microscopically, WT with classic triphasic components (blastemal, stromal, and epithelial cell types) are the most common (~40%); however, when a component predominates and accounts for more than two-thirds of the tumor, it is then classified accordingly. The blastema-predominant variant is the most common, and the stroma-predominant one is the rarest. Extensive sampling is required (at least one tissue section per cm of the largest dimension of the tumor) in order to characterize WT. The undifferentiated mesonephric blastemal cells are “small blue cells” with high nuclear-to-cytoplasmic ratios, coarse chromatin, and brisk mitotic activity. The blastemal component may be arranged in several patterns (including nodular, serpentine, and basaloid) and may be variably associated with a myxoid mesenchymal background.
In the common nephrogenic epithelial cell type, primitive columnar to cuboidal cells are arranged in glomeruloid and/or tubular structures. The stroma can be myxoid, with cells exhibiting fibrocystic or skeletal muscle differentiation. Heterologous differentiation can also be seen in any of the three components. The presence of anaplasia (defined as a threefold increase in nuclear diameter, hyperchromasia of the enlarged nuclei, and multipolar mitotic figures) is identified in less than 10% of WT and indicates “unfavorable histology” that correlates with a worse prognosis.
Immunohistochemical (IHC) stains may assist in establishing the diagnosis of WT depending on the tumor components. Positive nuclear immunostaining for WT1 is noted in close to 80% of blastema and primitive epithelial elements, but it may be negative in other tumor components.
The genetic alterations associated with WT are numerous and complex and include abnormalities (eg, mutations, deletions) of WT1, CTNNB1, AMER1 [WTX], and TP53; other reported alterations include loss of imprinting on 11p15 and loss of heterozygosity of both chromosomes 1p and 16q. These genetic alterations affect many aspects of tumor formation, disease progression, and response to therapy, among others. Testing for their presence or absence can assist in the development of a comprehensive treatment plan and the determination of significant prognostic factors.
Patients with WT are cured by initial therapy in close to 80% of cases. The presence of anaplasia and the identification of some genetic abnormalities have prognostic significance, and a low stage at the time of resection confers a “favorable” prognosis in WT. The majority of blastema-predominant WT usually respond to chemotherapy. Nevertheless, according to the SIOP-based literature, post-chemotherapy WT specimens with a predominance of viable blastemal tissue are considered high-risk tumors and are associated with a poor outcome.
Clear cell sarcoma of kidney (CCSK) is a rare tumor with an age range at presentation similar to that of WT. The malignant cells are polygonal with indistinct cell borders and round to oval nuclei with fine chromatin and small inconspicuous nucleoli. In the classic architectural pattern, they are arranged in nests or cords separated by arborizing fibrovascular septae. Other architectural patterns may be present and appear reminiscent of blastema-predominant WT. The genetic profile includes in-frame internal tandem duplication in the BCOR gene and t(10;17) resulting in YWHAE::NUTM2 gene fusion.
Primary intrarenal neuroblastomas are extremely rare tumors, so invasion of the kidney from an adjacent primary adrenal gland tumor needs to be excluded. Neuroblastomas are composed of small round to oval neoplastic cells with scanty cytoplasm, round to oval nuclei with “salt-and-pepper” chromatin, and poorly defined cell borders. The neoplastic cells are not uncommonly arranged in a lobular pattern associated with fine fibrovascular septae but may also be arranged in solid sheets. The background of the cellular process is variably composed of neuropil; Homer Wright rosettes may be seen. Tumor cells are immunopositive for PHOX2B and neuroendocrine markers (eg, CD56, synaptophysin, chromogranin, and neuron-specific enolase) and immunonegative for WT1.
Congenital mesoblastic nephroma is the most common renal tumor in patients under 1 year of age and is usually diagnosed during the first 3 months of life. It presents as a single, unilateral, and non-encapsulated renal tumor that tends to arise centrally (near the renal hilum). Three microscopic patterns (or variants) are recognized: classic, cellular, and mixed. Of these patterns, the cellular and mixed ones can be in the differential diagnosis of WT. In the cellular pattern (66% of cases), the cells are spindle-shaped or polygonal with high nuclear-to-cytoplasmic ratios and may be arranged in a herringbone pattern. It is morphologically identical to infantile fibrosarcoma and shares the chromosomal translocation t(12;15)(p13;q25), resulting in ETV6::NTRK3 fusion gene rearrangement. In the mixed pattern (10% of cases), areas with features of classic and cellular mesoblastic nephroma alternate.
Rhabdoid tumor of kidney (RTK) is a highly malignant tumor occurring in infants and young children. The malignant cells are polygonal and medium to large, with abundant eosinophilic cytoplasm, round nuclei with thick nuclear membranes, and large nucleoli; they are commonly arranged in a diffuse pattern. It is not uncommon for the cytoplasm to exhibit large eosinophilic inclusions that may displace the nuclei. Rhabdoid tumors exhibit mutation or deletion of SMARCB1 (also referred to as INI1, SNF5, and BAF47) located at chromosome 22q11 and loss of INI1 expression by immunohistochemistry.
Supplementary Questions
- Which of the following is true regarding blastema-predominant Wilms tumor?
- It is the most common type of congenital Wilms tumor in preterm patients
- Most blastema-predominant Wilms tumors exhibit SMARCB1 gene mutation or deletion
- The blastema-predominant Wilms tumor variant is the most common
- The blastemal component is always immunoreactive for synaptophysin and chromogranin
- The blastemal component is characterized by primitive columnar to cuboidal cells arranged in glomeruloid and/or tubular structures
- In the context of Wilms tumors, “anaplasia” is defined as which of the following?
- A relative increase in nuclear diameter when compared to the mesonephric blastemal cells, hyperchromasia of the enlarged nuclei, and multipolar mitotic figures
- A threefold increase in nuclear diameter, hyperchromasia of the enlarged nuclei, and multipolar mitotic figures
- Hyperchromasia of the enlarged nuclei, high nuclear to cytoplasmic ratio, and multipolar mitotic figures
- Increased number of atypical mitotic figures, nuclear hyperchromasia, and multinucleation
- More than 10 mitoses per 10 high-power fields and individual cell necrosis
- A 2-year-old girl has recently been diagnosed with a Wilms tumor. Her presenting signs and symptoms most likely included which of the following?
- Constipation
- Diabetes insipidus
- Enlarged abdomen
- Fever
- Lower body paralysis
References
- Delahunt B, Grignon DJ, Eble JN. Ch2. Tumors of the kidney. In: Amin MB, Grignon DJ, Srigley JR, Eble JN, eds. Urological Pathology. 1st ed. Lippincott Williams & Watkins. 2014:126-142.
- El Demellawy D, Cundiff CA, Nasr A, et al. Congenital mesoblastic nephroma: a study of 19 cases using immunohistochemistry and ETV6-NTRK3 fusion gene rearrangement. Pathology. 2016;48(1):47-50.
- Finn LS, Husain AN. Ch. 17, The Kidney and Lower Urinary Tract. In: Stocker JT, Dehner LP, Husain AN, eds. Stocker & Dehner’s Pediatric Pathology. Fourth ed. Wolters Kluwer. 2016;840-850.
- Goldblum JR, Folpe AL, Weiss SW. Ch.20 Rhabdomyosarcoma and Ch.33 Malignant Soft Tissue Tumors of Uncertain Type. In: Enzinger & Weiss’s Soft Tissue Tumors. 6th ed. Elsevier Saunders. 2014;601-638,1028-1045.
- WHO Classification of Tumours Editorial Board. Urinary and Male Genital Tumours. Lyon (France): International Agency for Research on Cancer. 2022. (WHO classification of tumours series, 5th Ed.; vol. 8).
- Shao L, Hill DA, Perlman EJ. Expression of WT-1, Bcl-2, and CD34 by primary renal spindle cell tumors in children. Pediatr Dev Pathol. 2004;7(6):577-582.
- van den Heuvel-Eibrink MM, van Tinteren H, Bergeron C, et al. Outcome of localised blastemal-type Wilms tumour patients treated according to intensified treatment in the SIOP WT 2001 protocol, a report of the SIOP Renal Tumour Study Group (SIOP-RTSG). Eur J Cancer. 2015;51(4):498-506.
Author
Nilsa C. Ramirez, MD, FCAP
Surgical Pathology Committee
Nationwide Children’s Hospital
Columbus, OH
Answer Key
- The blastema-predominant Wilms tumor variant is the most common (c)
- A threefold increase in nuclear diameter, hyperchromasia of the enlarged nuclei, and multipolar mitotic figures (b)
- Enlarged abdomen (c)