Clinical Summary
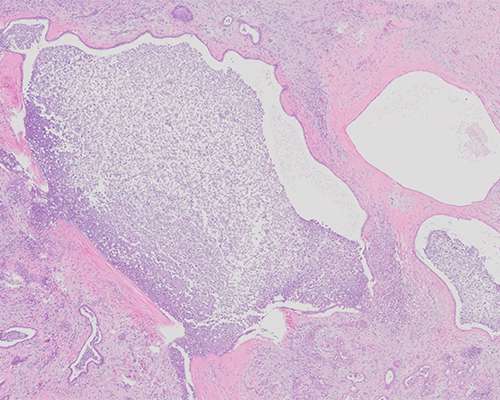
A 63-year-old woman undergoes orthotopic liver transplantation. She has a history of liver disease first diagnosed 25 years ago when she presented with hypertension and chronic renal failure due to polycystic kidney disease. Her explanted native liver weighs 4,943 grams and demonstrates multiple cysts visible over the capsular and cut surfaces. Many of these cysts contain straw-colored fluid and range in size from sub-centimeter to as much as 7 cm in greatest diameter and largely demonstrate smooth inner wall lining. Few cysts contain debris and/or clotted blood. The intervening liver parenchyma shows bands of fibrosis as well as tan-brown relatively preserved parenchyma.
Master List of Diagnoses
- Caroli disease
- Choledochal cyst
- Congenital hepatic fibrosis
- Mucinous cystic neoplasm (biliary cystadenoma)
- Polycystic disease of the liver
Archive Case and Diagnosis
This case first appeared as Performance Improvement Program in Surgical Pathology (PIP) 2018, Case 11, and is polycystic disease of the liver.
Criteria for Diagnosis and Comments
The histological sections show multiple cysts of varying sizes. The cysts are lined by flattened to cuboidal biliary-type epithelial cells surrounded by fibrous stroma. In some sections cysts with luminal old hemorrhages are present but no solid areas of epithelial proliferation are seen in any of the sections. Islands of compressed liver parenchyma are seen between these cysts as are scattered von Meyenburg complexes (bile duct hamartomas). Areas of parenchymal infarction secondary to chronic compression are seen in some areas. The findings are consistent with a diagnosis of polycystic disease of the liver.
Polycystic disease of the liver (PCDL) belongs to a group of inherited liver diseases collectively called fibropolycystic diseases of the liver. These are a heterogeneous group of congenital hepatobiliary disorders that include Caroli disease (CD), congenital hepatic fibrosis (CHF), and PCDL. The most common presenting symptoms of PCDL are abdominal mass effect from liver enlargement and pain secondary to hemorrhages or infections. PCDL presents in adults and has a wide age of onset, but is rarely in teenagers, with females having earlier presentation and generally a more severe disease. The main clinical problems of PCDL are caused by the large size of the affected liver and include abdominal distention, shortness of breath, and pain secondary to intra-cystic hemorrhage, infection, or tension on the Glisson (liver) capsule. The risk factors for disease progression include a family member with the disease, young age at initial diagnosis, female sex, exogenous estrogen use, and multiple pregnancies. Liver function is usually preserved but patients could occasionally develop portal hypertension due to liver cysts impingement on portal vein inflow and/or obstruction to hepatic vein drainage. Risk for carcinoma developing in the cysts exists but very low.
Radiologic diagnosis of PCDL is rather straight forward and can be achieved by ultrasound (USS), magnetic resonance imaging (MRI), or computed tomography (CT) scans. Ultrasound examination shows hepatomegaly with cysts that could range from localized to diffuse; the cysts appear anechoic and homogeneous. MRI provides superior resolution to USS or CT and is preferred when necessary. Treatment options are aimed at reducing liver mass and include abdominal percutaneous puncture, laparoscopic or open fenestration, surgical resection, or transplantation. Long-term outcome for transplantation after the first two months is excellent, with most of the complications occurring in the immediate post-transplant days to weeks.
Gross examination of liver in PCDL reveals massive hepatomegaly with the liver weighing up to 10 kilograms in the most extreme cases. The liver contains multiple cysts that range from small to large, the larger cysts measuring 10 cm or more in size. The presence of these cysts invariably leads to alterations in the normal liver shape. The PCDL cysts are non-communicating with the functioning portions of the biliary system, unlike the cysts in CD. Histologic examination demonstrates cysts lined by single biliary cuboidal to simple columnar epithelium. Flattened or sloughed off epithelium could be seen as a result of long-standing distention, hemorrhage, or other degenerative changes. Over several years there is progressive enlargement of these cysts scattered throughout the liver parenchyma, leading in parallel to increasing fibrosis of the surrounding parenchyma. In addition to large cysts, patients could have other evidence of ductal plate malformation characterized by multiple small aggregates of cysts clustered into the entity referred to as von Meyenburg’s complexes or bile duct hamartomas (or micro-hamartomas).
PCDL occurs in two distinct genetic contexts; the first is associated with the autosomal dominant polycystic kidney disease (AD-PCKD) and the other is referred to as autosomal dominant PCDL (AD-PCDL) or isolated PCDL, which is not associated with renal disease. In the former, PCDL is the commonest extra-renal manifestation of the renal syndrome, occurring in up 90% of AD-PCKD patients; less commonly pancreatic cysts and cerebral aneurysms could also manifest as part of the syndrome. The genetic basis for AD-PCKD is a mutation in the PKD1 or PKD2 gene on chromosomes 16p and 4q21 respectively. The protein products of these genes, polycystin-1 and polycystin-2, when altered lead to dysregulation of fluid secretion and growth of cyst lining epithelium. Patients present earlier with kidney-related symptoms including progressive renal failure, with liver function often reasonably preserved until much later in life and few patients requiring liver transplantation. The second genetic variant, AD-PCDL, results from mutation in the PRKCSH, (that encodes hepatocystin), or the SEC63, (that encodes Sec63 protein) genes on chromosome 19p or 6q21, respectively, and accounts for 33% to 50% of all cases of PCDL. Hepatocystin and Sec63, products of the mutated genes, alter the functions of polycystin-1 and polycystin-2, which explains the similar phenotypes between the two genetic variants. Recently, another gene is identified, the LRP5 (Low density lipoprotein Receptor-related Protein 5) gene as the third locus associated with isolated PCDL by whole-exome sequencing in an extended family.
CD is an autosomal recessive, congenital, communicating, multifocal, cystic dilatation of the large intrahepatic bile duct. It is associated with the autosomal recessive PCKD (AR-PCKD). Symptomatic patients develop biliary colic, recurrent cholangitis, intrahepatic lithiases, and cholestasis with an increased risk for cholangiocarcinoma. CD presents much earlier than PCDL, typically in childhood or early adulthood with jaundice, abdominal pain, and other complications of calculi and infections. When CD co-exists with CHF, it is regarded as the Caroli syndrome. CHF and CD are therefore best regarded as a continuum of the same disease, differentiated by the level where the bile duct plate arrest occurs. CHF affects mainly the small intrahepatic bile ducts causing abnormal bile duct proliferation in malformed portal areas and fibrosis. Portal and periportal fibrosis is variable and dilated, tortuous ducts with irregular contours with or without inspissated bile are seen in the periphery of portal triads. CHF could occur alone, presenting very early in childhood as portal hypertension and in association with AR-PCKD, while CD is less commonly isolated, more likely occurring as the Caroli syndrome (ie, in association with the CHF). A less common association of CHF with the AD-PCKD has also been recognized.
Choledochal cyst is abnormal dilatation of the large biliary tree classified into five types depending on the site of involvement. The first four types affect perihilar or distal extrahepatic bile ducts while type 5 is intra-hepatic and corresponds to CD. The commonest type is type 1, characterized by partial or complete saccular or fusiform dilatation of the common bile duct, without intrahepatic involvement. The presentation could be in infancy, childhood, adolescence, or adulthood. In infants, an abdominal mass can be felt in addition to jaundice, acholic stool, and hepatomegaly. In adults, presentation is usually from complications of calculi and/or infection and could range from vague abdominal discomfort to more severe pain, fever, and jaundice. Less commonly, pancreatitis could occur especially in those with type 3 (choledochocele) involving distal end, intra-duodenal portion, of the common bile duct.
Unlike all other entities discussed above, mucinous cystic neoplasm (MCN), formerly called biliary cystadenoma is neoplastic in nature, but accounts for less than 5% of intrahepatic cystic lesions. MCN is a benign cyst that arises mainly from the intrahepatic ducts and rarely from the extrahepatic ducts or gallbladder. It most frequently consists of a single, sometimes multiloculated cyst, with predilection for right lobe of the liver. MCN occurs almost exclusively in women and has a high rate of recurrence and a potential for malignant transformation. MCN is lined by simple epithelium with varying degree of mucin prominence. The underlying stroma is ovarian-type, and the stromal cells express progesterone receptor by immunohistochemistry.
Supplementary Questions
- Which of the following liver cysts results from mutations in the PRKCSH gene that encodes hepatocystin?
- Caroli disease
- Choledochal cyst
- Congenital hepatic fibrosis
- Mucinous cystic neoplasm (biliary cystadenoma)
- Polycystic disease of the liver
- Which of the following conditions is most likely to present with pancreatitis?
- Caroli disease
- Choledochal cyst
- Congenital hepatic fibrosis
- Mucinous cystic neoplasm (biliary cystadenoma)
- Polycystic disease of the liver
- Which of the following conditions is associated with autosomal recessive polycystic kidney disease?
- Cholangiocarcinoma
- Choledochal cyst
- Congenital hepatic fibrosis
- Mucinous cystic neoplasm (biliary cystadenoma)
- Polycystic disease of the liver
References
- Abu-Wasel B, Walsh C, Keough V, Molinari M. Pathophysiology, epidemiology, classification and treatment options for polycystic liver diseases. World J Gastroenterol. 2013;19(35):5775–5786.
- Cnossen WR, Drenth JP. Polycystic liver disease: an overview of pathogenesis, clinical manifestations and management. Orphanet J Rare Dis. 2014;9:69-81.
- Everson GT. Polycystic Liver Disease. Gastroenterol Hepatol (NY). 2008;4(3):179–181.
- O'Brien K, Font-Montgomery E, Lukose L, et al. Congenital hepatic fibrosis and portal hypertension in autosomal dominant polycystic kidney disease. J Pediatr Gastroenterol Nutr. 2012;54(1):83-89.
- Pirson Y. Extrarenal manifestations of autosomal dominant polycystic kidney disease. Adv Chronic Kidney Dis. 2010;17(2):173-180.
- Todani T, Watanabe Y, Narusue M, Tabuchi K, Okajima K. Congenital bile duct cysts: Classification, operative procedures, and review of thirty-seven cases including cancer arising from choledochal cyst. Am J Surg. 1977;134(2):263-269.
Authors
Nawal Al-Mohammadi, MD
Liver and Hepatobiliary Pathology Fellow
University Health Network/University of Toronto
Toronto, ON
Oyedele A. Adeyi, MB, BS
Surgical Pathology Committee
University Health Network/ University of Toronto
Toronto, ON
Answer Key
- Polycystic disease of the liver (e)
- Choledochal cyst (b)
- Congenital hepatic fibrosis (c)