Clinical Summary
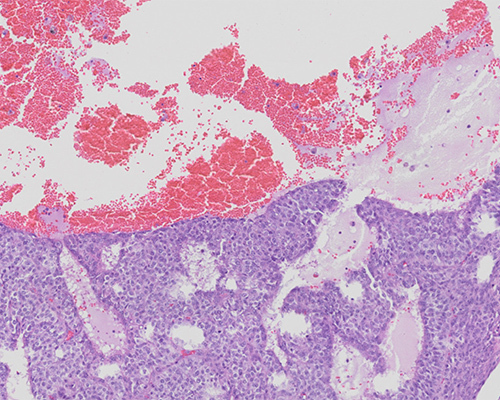
A 9-year-old girl presents to her pediatrician with premenarchal Tanner stage II-III breast development, mildly accelerated growth for chronological age, and vague abdominal pain. Physical examination demonstrates a palpable pelvic mass, and ultrasound confirms a 9.0 cm solid/cystic right ovarian mass. Ultrasound also shows an enlarged uterus, consistent with precocious puberty. Laboratory evaluation reveals notable elevations in serum estradiol and inhibin B levels for her age, with low serum FSH and LH. CA125 is normal. Following oophorectomy, macroscopic examination demonstrates a yellow-tan 9.5 cm solid and cystic ovarian mass with focal areas of hemorrhage.
Master List of Diagnoses
- Adult granulosa cell tumor
- Juvenile granulosa cell tumor
- Malignant melanoma of the ovary
- Small cell carcinoma of the ovary, of hypercalcemic type
- Yolk sac tumor
Archive Case and Diagnosis
This case first appeared as Performance Improvement Program in Surgical Pathology (PIP) 2020, Case 34, and is juvenile granulosa cell tumor of the ovary.
The information provided in this case was accurate and correct at the time of publication in 2020.
Any changes in terminology since the time of publication may not be reflected in this case.
Criteria for Diagnosis and Comments
The patient presented with signs of isosexual precocious puberty, abdominal pain, and a palpable ovarian mass with elevated serum estradiol and inhibin B levels. These features in this age group are strongly suggestive of a juvenile granulosa cell tumor (JGCT). Microscopic examination shows solid areas and follicle-like cystic areas. The follicles contain watery, basophilic fluid and detached tumor cells admixed with pigment-laden macrophages. The solid areas are composed of cells with rounded nuclei with moderate nuclear pleomorphism and moderate amounts of eosinophilic cytoplasm. Scattered mitoses are noted. Nuclear grooves and/or Call–Exner bodies are not readily identified. Immunostains show diffuse strong staining for inhibin, calretinin, WT1, and SF1. Stains for CK7 and HMB45 were negative and staining for SMARCA4/BRG1 is retained.
Granulosa cell tumors (GCT) constitute fewer than 5% of all ovarian tumors but represent approximately 70% of ovarian sex cord-stromal tumors. In the United States, the annual incidence of all GCT diagnoses ranges between 0.4 - 1.7 cases per 100,000 women. GCT arise from the somatic cells of the sex cords and are divided into two distinct subtypes—adult granulosa cell tumors (AGCT) and JGCT. AGCT are usually identified in peri- and post-menopausal women; are often detected at an early stage due to accompanying symptoms of abdominal pain, post-menopausal bleeding, and/or hyperestrogenism; and usually follow an indolent course. These can be associated with endometrial hyperplasia or carcinoma. JGCT is very rare and presents in pre- and peri-menarchal women; approximately 90% of GCT in women less than 30 years of age are of this type. Importantly, adult and juvenile granulosa cell tumors also show differing molecular profiles, which suggests that they are truly distinct tumors.
Juvenile granulosa cell tumors usually present clinically with isosexual precocious puberty due to elevated estradiol levels. The triad of a palpable adnexal mass, elevated serum estradiol and decreased gonadotropin (FSH and LH) levels are characteristic of JGCT in a premenarchial woman. Some JGCT also secrete progesterone and/or testosterone, which can cause dramatic virilization in young women. Serum inhibin B levels are often elevated and can serve as a useful serum tumor marker to monitor for recurrence with a reported sensitivity of 89% and a specificity of 100%.
Approximately 90% of JGCT are low stage (stage IA or IB) at diagnosis. Tumors can range from 3 - 35 cm in diameter, with an average size of 12.5 cm. Macroscopically, JGCT show similar features to AGCT, with solid and cystic growth patterns. Hemorrhage and/or necrosis may be present. The microscopic appearance is characterized by a diffuse arrangement of neoplastic cells with pleomorphic and hyperchromatic nuclei with abundant cytoplasm. Follicle formation can be prominent in JGCT, but the characteristic features of AGCT, such as Call–Exner bodies and grooved nuclei, are uncommon and focal, if present.
Surgical resection is the primary treatment for JGCT. However, due to the young age of most patients, the extent of surgery must be carefully evaluated, and many surgeons prefer to perform unilateral oophorectomy to preserve fertility. Staging in these young patients includes exploratory laparotomy and peritoneal cytology, with removal of all visible disease. Prognostic factors include stage, size, degree of nuclear atypia, and mitotic activity. Interestingly, tumor rupture (which occurs in 10% of cases) does not seem to negatively impact prognosis. Survival for stage I JGCT is 78%-92%. However, recurrences in JGCT tend to have a worse prognosis and may progress rapidly; thus, clinical monitoring for an extended period is recommended utilizing serum studies for estradiol, inhibin B, and imaging modalities.
Many JGCT show aneuploidy, trisomy 12 being common. FOXL2 immunostains are positive in almost 95% of all granulosa cell tumors (adult and juvenile types). Immunopositivity for FOXL2 and SF1 is considered to indicate sex cord-stromal tumor differentiation. These markers also stain other sex cord-stromal tumors, including fibromas, steroid cell tumors, and sclerosing stromal tumors without FOXL2 mutations. Positive FOXL2 staining, however, does not imply the presence of a FOXL2 mutation. When present, the FOXL2 402C>G point mutation is specific for AGCT and an important distinguishing feature from other sex-cord stroma tumors. Few genetic syndromes have been associated with JGCT, including Maffucci syndrome (enchondromas, hemangiomas, phleboliths) and Ollier disease (enchondromatosis).
Adult granulosa cell tumors can generally be distinguished from JGCT based on their unique clinical age-profile at presentation. However, it should be noted that either type can occur in any age group, albeit uncommonly. The histology of JGCT also departs from AGCT by showing variably sized follicle-like spaces, a lack of conspicuous Call–Exner bodies, nuclei lacking noticeable grooves, more abundant cytoplasm, and increased cytologic atypia. Immunostains, including FOXL2, will show significant overlap between these entities, but lesions with FOXL2 mutation should be classified as AGCT.
Malignant melanoma of the ovary can present with unilateral or bilateral ovarian mass(es) but is not estrogenic. Microscopically, ovarian melanoma may show a variety of patterns, including pseudo-follicular and nested. However, one may also appreciate intracellular melanin pigment, and the presence of bilateral disease usually argues against JGCT. When necessary, immunostains can be helpful in the differential with JGCT, with typical immunopositivity seen for HMB45, S100, and SOX10 in melanoma. Melanomas are also negative for inhibin, SF1, and FOXL2 immunostains. However, Melan-A should be interpreted with caution in this differential, as it can be positive in some sex cord-stromal tumors.
Small cell carcinoma of the ovary, of hypercalcemic type (SCCOHT) is included in the differential diagnosis with JGCT due to overlapping age distributions and presentation with a unilateral ovarian mass. SCCOHT typically occur in women over 20; while this is older than the typical JGCT age at presentation of 13 years, there can be some age overlap in these two tumors. SCCOHT typically present with hypercalcemia and lack estrogenic manifestations clinically. Microscopically, these tumors consist of homogenous cells with hyperchromatic nuclei, but occasional follicle-like spaces can be seen. It is crucial to correctly distinguish these entities due to the poor prognosis associated with SCCOHT. If necessary, immunostains may differentiate these entities more definitively via negative inhibin and calretinin, and loss of SMARCA4/BRG1 immunoexpression in SCCOHT.
Yolk sac tumor, or endodermal sinus tumor, is a malignant germ cell tumor. These tumors originate within the first through fourth decades, with a median age at diagnosis of 19 years. Thus, these tumors show overlapping age ranges with JGCT, and patients present similarly with abdominal pain and a pelvic mass. However, yolk sac tumors are generally not estrogenic, and patients instead have markedly elevated serum alpha-fetoprotein levels, which are either normal or, rarely, elevated in JGCT. Yolk sac tumor can also display a variety of histologic and cytologic patterns, some of which can show partial overlap with JGCT. However, the presence of diagnostic Schiller-Duval bodies and positive SALL4 immunoreactivity, as well as negative inhibin, SF1, and FOXL2 immunostains, in yolk sac tumor easily separates these entities.
Supplementary Questions
- In a premenarchal girl, which triad is strongly indicative of juvenile granulosa cell tumors (JGCT)?
- Elevated insulin levels, bilateral ovarian cysts, and hirsutism
- Elevated serum estradiol, decreased serum FSH, and pelvic mass
- Enchondromas, mucosal pigmentation, and spider angiomas
- Pelvic mass, elevated serum prolactin, and galactorrhea
- Pelvic mass, skin tags, and pituitary adenoma
- Which of the following is true regarding JGCT?
- Aneuploidy is rare.
- FOXL2 immunopositivity is diagnostic.
- FOXL2 immunopositivity may be seen in the absence of FOXL2 mutation.
- FOXL2 mutation (402C>G) is pathognomonic.
- Inhibin immunostain is negative.
- JGCT typically demonstrate which of the following findings?
- Bilateral ovarian involvement, cellular pigmentation, and SOX10 immunoreactivity
- Cystic and solid patterns, inhibin negativity, and positive SALL4 immunoreactivity
- Cystic and solid patterns, inhibin positivity, and SF1 immunoreactivity
- Hyperchromatic nuclei, hypercalcemia, and loss of BRG1
- Nuclear grooves, Call–Exner bodies, and FOXL2 mutation
References
- Al-Agha OM, Huwait HF, Chow C, et al. FOXL2 is a sensitive and specific marker for sex cord-stromal tumors of the ovary. Am J Surg Pathol. 2011;35(4):484-494.
- D’Angelo E, Mozos A, Nakayama D, et al. Prognostic significance of FOXL2 mutation and mRNA expression in adult and juvenile granulosa cell tumors of the ovary. Mod Pathol. 2011;24(10):1360-1367.
- Kao CS, Cornejo KM, Ulbright TM, Young RH. Juvenile granulosa cell tumors of the testis: A clinicopathologic study of 70 cases with emphasis on its wide morphologic spectrum. Am J of Surg Pathol. 2015;39(9);1159-1169.
- Schultz KP, Harris AK, Finch M, et al. DICER1-related Sertoli-Leydig cell tumor and gynandroblastoma: Clinical and genetic findings from the International Ovarian and Testicular Stromal Tumor Registry. Gynecol Oncol. 2017;147(3)521-527.
- Sivasankaran S, Itam P, Ayensu-Coker L, et al. Juvenile Granulosa Cell Ovarian Tumor: A Case Report and Review of Literature. J Pediatr Adolesc Gynecol. 2009;22(5):e114-e117.
- Young RH, Dickersin GR, Scully RE. Juvenile granulosa cell tumor of the ovary A Clinicopathological Analysis of 125 Cases. Am J Surg Pathol. 1984;8(8):575-596.
Answer Key
- Elevated serum estradiol, decreased serum FSH, and pelvic mass (b)
- FOXL2 immunopositivity may be seen in the absence of FOXL2 mutation. (c)
- Cystic and solid patterns, inhibin positivity, and SF1 immunoreactivity (c)