- Home
- Member Resources
- Pathology Case Challenge
- Retroperitoneum
Clinical Summary
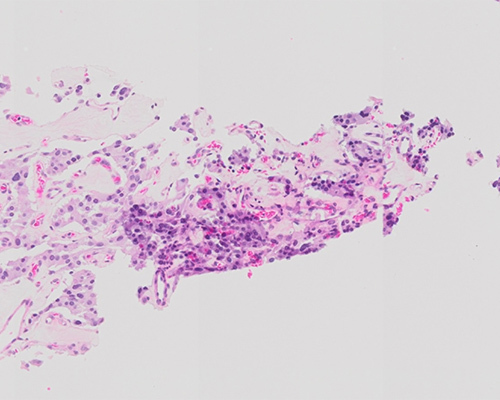
A 52-year-old man presents with abdominal pain radiating to his left lower back. Except for minor weakness of the lower extremities, there are no other symptoms. Computed tomography scan of the chest, abdomen, and pelvis reveals a well-defined 7 cm lesion located in the retroperitoneum, with scattered intratumoral calcifications. Imaging also reveals enlarged mesenteric and inguinal lymph nodes. A total body positron emission tomography scan demonstrates increased uptake of fluorodeoxyglucose within the retroperitoneal lesion and lymph nodes. A surgical resection is performed.
Master List of Diagnoses
- Adrenal cortical adenoma
- Alveolar soft part sarcoma
- Gastrointestinal stromal tumor
- Malignant paraganglioma
- Melanoma
- Renal cell carcinoma
- Well-differentiated neuroendocrine tumor (carcinoid)
Archive Case and Diagnosis
This case first appeared as Performance Improvement Program in Surgical Pathology (PIP) 2019, Case 27, and is malignant paraganglioma in the retroperitoneum.
Criteria for Diagnosis and Comments
Sections show a well-circumscribed mass with focal areas of hemorrhage, consisting of round epithelioid cells arranged in compact cell nests and trabeculae, surrounded by spindled cells and fibrovascular tissue. Tumor cells generally have centrally located nuclei with fine chromatin and a moderate amount of eosinophilic, granular cytoplasm. The mass is moderately to highly cellular with focal areas of profound nuclear pleomorphism and nuclear hyperchromasia. Focal lymphovascular invasion is identified. No significant necrosis or increased mitotic rate is identified. Lymph nodes showed involvement by the tumor as well. These findings are consistent with the diagnosis of a malignant paraganglioma.
Paragangliomas are highly vascular tumors with a fleshy, pink to red-brown to gray gross appearance, due to hemorrhage or fibrosis. They typically have a thin capsule and are classically composed of round epithelioid cells arranged in compact cell nests or “zellballen.” Tumor cells generally have centrally located nuclei with finely clumped chromatin and a moderate amount of eosinophilic, granular cytoplasm. The classic immunohistochemical profile of the tumor is positivity for neuron-specific enolase, synaptophysin, and/or chromogranin, and negativity for cytokeratins (CK), desmin, SMA, and TFE3. The peripheral sustentacular cells usually stain positive for S100 and negative for neuroendocrine markers.
Approximately 500 to 1600 cases of paraganglioma occur in the United States each year. Paragangliomas are non epithelial neuroendocrine tumors that arise from the extra-adrenal autonomic paraganglia, which are derived from the embryonic neural crest. They are closely related to pheochromocytomas (intra-adrenal paragangliomas), which have the potential to behave in an aggressive fashion. The Pheochromocytoma of the Adrenal Gland Scales Score (PASS) score evaluates twelve histological features to aid in predicting behavior of paraganglioma. These features and the PASS score for this case are provided in the table below.
PASS scoring scale:
FEATURE | Score if present (points assigned) | Score for this case |
Large nests or diffuse growth (>10% of tumor volume) | 2 | 0 |
Central (medium/large nests) or confluent tumor necrosis | 2 | 0 |
High cellularity | 2 | 2 |
Cellular monotony | 2 | 0 |
Tumor cell spindling (even if focal) | 2 | 0 |
Mitotic figures >3/10 high-power fields | 2 | 0 |
Atypical mitotic figures | 2 | 0 |
Extension into adipose tissue | 2 | 0 |
Vascular invasion | 1 | 1 |
Capsular invasion | 1 | 0 |
Profound nuclear polymorphism | 1 | 1 |
Nuclear hyperchromasia | 1 | 1 |
TOTAL | 20 | 5 |
A PASS score less than 4 suggests a benign indolent lesion, and values greater than 6 suggest a tumor with malignant potential. The tumor described in this case had a PASS score of 5, intermediate between benign and malignant. According to 2004 World Health Organization criteria, however, malignancy is determined by the presence of metastatic spread, and therefore the paraganglioma in this case would be defined as malignant regardless of PASS score, as it was metastatic to lymph nodes.
Extra-adrenal paragangliomas derived from sympathetic paravertebral ganglia are usually located within the abdomen (75%), bladder/prostate (10%), or base of the skull (5%). Most sympathetic paragangliomas secrete catecholamines and are malignant in 25%-60% of cases. Parasympathetic paragangliomas are often located along the glossopharyngeal and vagal nerves in the neck and base of the skull, most commonly in the carotid body, and are usually nonfunctional and less likely to be malignant. Paragangliomas in the central nervous system usually present as a well-circumscribed mass in the filum terminale. Gangliocytic paraganglioma is a rare tumor typically found in the duodenum, which may be sporadic or arise in the setting of neurofibromatosis type 1.
While most paragangliomas are sporadic, approximately 30%-50% are associated with an inherited syndrome. A subset of hereditary paragangliomas have been linked to mutations in genes of the succinate dehydrogenase (SDH) enzyme complex, multiple endocrine neoplasia types 2A and 2B (MEN2), neurofibromatosis type 1 (NF1), von Hippel–Lindau (VHL), and Carney–Stratakis syndrome. For other syndromes and disease associations such as the Carney triad (characterized by gastric gastrointestinal stromal tumor, pulmonary chondroma, and extra-adrenal paraganglioma, mainly in young women), a specific genetic defect has not been identified.
Adrenal cortical adenoma typically presents as a well-circumscribed lesion with a yellow-brown cut surface. Several architectural patterns exist, but this entity can have a nested zellballen architecture and stain positive for synaptophysin and negative for CKs, like paragangliomas. However, unlike paragangliomas, adrenal cortical adenomas often contain clear lipid-rich cells typical of the adrenal cortex. In contrast to paragangliomas, these cells are typically chromogranin negative, and positive for adrenocortical markers such as SF1, inhibin A, and Melan-A.
Alveolar soft part sarcoma (ASPS) is typically found in deep soft tissue and consists of large polygonal cells with abundant eosinophilic cytoplasm. In contrast to paragangliomas, ASPS usually stains negative for synaptophysin, chromogranin, and S100 protein, and positive for nuclear TFE3. It exhibits the t(X;17)(p11;q25) ASPSCR1::TFE3 gene fusion.
Well-differentiated neuroendocrine tumors (carcinoids) can also present as a well-circumscribed lesion with a similar nested architecture appearance as paraganglioma. They are often synaptophysin positive. However, the tumor cells have more of a speckled “salt-and-pepper” nuclear chromatin pattern and are CK positive, and sustentacular cells are absent. They are also rare in the retroperitoneal area.
Gastrointestinal stromal tumor typically arises in the muscular wall of stomach and may present as a well-circumscribed lesion. However, unlike paraganglioma, the tumor cells often stain negative for synaptophysin and chromogranin, and positive for CD117 and DOG1.
Metastatic melanoma can involve the adrenal gland and can usually be distinguished from paraganglioma based on immunohistochemistry for synaptophysin and chromogranin (negative in melanoma, positive in paraganglioma) and S100, HMB45, and MART1 (positive in melanoma, negative in paraganglioma with positive S100 staining in sustentacular cells). Rare cases of melanin-expressing pigmented pheochromocytoma have been documented, but the cells of these tumors stain negative for melanocytic markers.
Renal cell carcinoma (RCC) may present as a “retroperitoneal mass” or metastasize to the adrenal glands. Grossly, the cut surface of clear cell RCC is golden-yellow, compared to the fleshy red brown-gray cut surface of a typical paraganglioma, although areas of hemorrhage are usually seen in both tumors. Clear cell RCC can have a nested architecture similar to paraganglioma, with nests surrounded by intricate, branching fibrovascular septations, but it can also exist as solid sheets. In contrast to paragangliomas, RCC has strong CK positivity and is also positive for EMA, CD10, RCC, and PAX8.
Supplementary Questions
- Which of the following statements regarding malignant paragangliomas is true?
- Definitive diagnosis of malignant paraganglioma requires identification of metastasis
- Parasympathetic paragangliomas are more often malignant than sympathetic paragangliomas
- They are defined by atypical mitoses
- They are identified by nuclear pleomorphism
- They are most common in the head and neck
- Which of the following statements regarding extra-adrenal paragangliomas is true?
- Gangliocytic paraganglioma usually arises in the stomach
- More than 80% of sympathetic paragangliomas are malignant
- Most (70% - 75%) extra-adrenal paragangliomas are related to inherited mutations
- Parasympathetic paragangliomas are common in the head and neck, often in association with nerves
- The Carney triad consists of paragangliomas, gastrointestinal stromal tumor of the stomach, and small cell neuroendocrine carcinoma of the lung
- Which of the following is a component of the PASS score?
- Fewer than 3 mitotic figures per 10 high-power fields
- Intracytoplasmic inclusions
- Large nests or diffuse growth (greater than 10% of tumor volume)
- Low cellularity
- Prominent degenerative change
References
- Burnichon N, Brière JJ, Libé R, et al. SDHA is a tumor suppressor gene causing paraganglioma. Hum Mol Genet. 2010;19(15):3011-3020.
- Chen H, Sippel RS, O'Dorisio MS, et al. The North American Neuroendocrine Tumor Society consensus guideline for the diagnosis and management of neuroendocrine tumors: pheochromocytoma, paraganglioma, and medullary thyroid cancer. Pancreas. 2010;39(6):775-783.
- de Wailly P, Oragano L, Radé F, et al. Malignant pheochromocytoma: new malignancy criteria. Langenbecks Arch Surg. 2012;397(2):239-246.
- Fishbein L, Merrill S, Fraker DL, et al. Inherited mutations in pheochromocytoma and paraganglioma: why all patients should be offered genetic testing. Ann Surg Oncol. 2013;20(5):1444-1450.
- Kakkar A, Kaur K, Kumar T, et al. Pigmented pheochromocytoma: an unusual variant of a common tumor. Endocr Pathol. 2016;27(1):42-45.
- Lee JA, Duh QY. Sporadic paraganglioma. World J Surg. 2008;32(5):683-687.
- Strong VE, Kennedy T, Al-Ahmadie H, et al. Prognostic indicators of malignancy in adrenal pheochromocytomas: clinical, histopathologic, and cell cycle/apoptosis gene expression analysis. Surgery. 2008;143(6):759-768.
- Tischler A.S. et al. WHO Classification of tumours of the adrenal medulla and extra-adrenal paraganglia. In Lloyd RV, Osamura RY, Kloppel G, Rosai J (eds). WHO classification of tumours: pathology and genetics of tumours of endocrine organs. 4th ed. Lyon: IARC. 2017;180-207.
- Thompson LD. Pheochromocytoma of the Adrenal gland Scaled Score (PASS) to separate benign from malignant neoplasms: a clinicopathologic and immunophenotypic study of 100 cases. Am J Surg Pathol. 2002;26(5):551-566.
Authors
Jake Maule, MD, PhD
Pathology Resident
Duke University
Durham, NC
Xiaoyin “Sara” Jiang, MD, FCAP
Surgical Pathology Committee
Duke University
Durham, NC
Answer Key
- Definitive diagnosis of malignant paraganglioma requires identification of metastasis (a)
- Parasympathetic paragangliomas are common in the head and neck, often in association with nerves (d)
- Large nests or diffuse growth (greater than 10% of tumor volume) (c)