Clinical Summary
A 36-year-old man presents to his primary care physician with complaints of an enlarging mass in his thigh. Physical exam demonstrates a large, firm, fusiform mass in the deep anterior thigh measuring approximately 10 x 7 x 6 cm. The patient does not recall any trauma to the area, and the mass is not painful to palpation. A needle biopsy and subsequent wide resection of this mass is performed. Macroscopic examination demonstrates a 10 cm mass with a gelatinous cut surface without any grossly identified areas of necrosis.
Master List of Diagnoses
- Dedifferentiated liposarcoma
- Myxofibrosarcoma
- Myxoid liposarcoma
- Low-grade fibromyxoid sarcoma
- Pleomorphic liposarcoma
Archive Case and Diagnosis
This case first appeared as Performance Improvement Program in Surgical Pathology (PIP) 2019, Case 12, and is a myxoid liposarcoma presenting in the thigh.
Criteria for Diagnosis and Comments
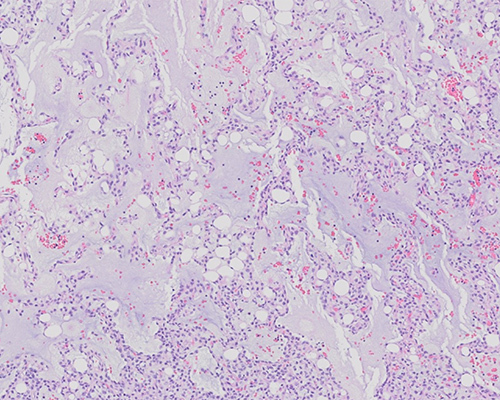
Histologic sections demonstrate a moderately cellular lesion composed of spindled and ovoid cells set within a myxoid stroma, with focal hemorrhage. Focal “pools” of myxoid material impart an almost pulmonary edema-like appearance. Scattered adipocytes are present, as are occasional vacuolated lipoblasts. A prominent plexiform and branching vascular pattern is noted, but mitoses are rare (fewer than 1 per high-power field). Immunostains demonstrate focal S100 immunopositivity, with negative MDM2, CDK4, and Melan-A immunostains. Fluorescence in situ hybridization (FISH) study for DDIT3 (CHOP) rearrangement is positive. This is consistent with a diagnosis of a myxoid liposarcoma (MLPS).
Liposarcomas are considered the most common soft tissue sarcomas and account for approximately 20% of all new adult sarcoma diagnoses. Liposarcomas are a heterogeneous group of tumors, which the World Health Organization has classified into subtypes based on distinct histologic and molecular differences, disease-free survival and overall survival. These subtypes include well-differentiated and dedifferentiated liposarcoma, myxoid liposarcoma (MLPS), and pleomorphic liposarcoma.
MLPS is the second most common subtype of liposarcoma, following well-differentiated liposarcoma, and accounts for approximately 30% of all liposarcomas. MLPS usually arises in younger adults and is the most frequent liposarcoma subtype in patients less than 20 years of age. Unlike well-differentiated liposarcoma, which commonly arises within the retroperitoneum, MLPS has a predilection for the deep soft tissues of the extremities and back. Indeed, the most common site of origin for MLPS is the thigh. MLPS, in contrast to other liposarcoma subtypes, also has a propensity to metastasize to unusual fat-bearing non-pulmonary sites, including bone, bone marrow, fatty areas in the mediastinum and retroperitoneum, and other visceral organs. Clinical followup for MLPS patients should therefore include skeletal surveys and magnetic resonance imaging (MRI) to monitor for bony and visceral organ metastases.
Histologically, MLPS is typified by a usually sparse mesenchymal spindle or oval cell component present within a prominent myxoid stroma. Uni- or bi-vacuolated or signet ring lipoblasts, mature adipocytes, and a distinctive plexiform vasculature are characteristic features. A round cell component in MLPS indicates a higher-grade histology with associated prognostic impact but is no longer considered as a separate “round cell liposarcoma” diagnostic category. Immunostains serve mostly to exclude other entities considered in the histologic diagnosis of MLPS. S100 immunostain may be positive, but MLPS is usually negative for keratins, CD34, SMA, desmin, and other soft tissue markers. MDM2 and CDK4 immunostains, characteristically positive in well-differentiated/dedifferentiated liposarcomas, are typically negative in MLPS.
Genetic and molecular tests are also used diagnostically for MLPS, as they harbor a diagnostic genetic abnormality of t(12;16)(q13.p11) in 95% of cases. This results in the abnormal fusion gene FUS::DDIT3 (also called FUS::CHOP in older literature), which results in fusion transcripts classified into 3 types: type I (exons 7-2), type II (exons 5-2), and type III (exons 8-2). While these 3 types of transcripts are known to play a role in MLPS oncogenesis, the exact relationship between the type of transcript and clinical outcome remains unknown. In addition, a different translocation, t(12;22)(q13;q12), has been rarely identified in a small subset of MLPS and results in a fusion rearrangement between EWS and DDIT3. This less frequent fusion also results in several different types of fusion transcripts: type I (exons 7-2), type II (20-2), type III (13-2), and type IV (exons 13-3), with type I thought to manifest the best clinical outcome.
Complete surgical excision is the mainstay of therapy for liposarcoma, including MLPS; however, MLPS recur locally in up to 30% of cases with surgery alone. Low-grade tumors metastasize in fewer than 10% of cases, but high-grade tumors (>5% round cells) have significantly increased risk of metastasis (up to 45%-60% of cases). Indeed, the 5-year overall survival of 90% for MLPS without a round cell component drops to approximately 50% for those rich in round cells. Therefore, the presence of a round cell component >5% is considered the most important prognostic factor in MLPS. Other adverse prognostic indicators include age >45 years, presence of coagulative necrosis, p53 overexpression, and TP53 and CDKN2A mutations.
Other treatment modalities for MLPS include radiation therapy, chemotherapy, and novel agents targeting MLPS specifically. MLPS, as opposed to other liposarcoma subtypes, is considered highly radiosensitive, and neoadjuvant radiation therapy often provides dramatic responses and yields an easier surgical excision. Indeed, for low-grade MLPS, local control rates of 97% have been reported for combined surgery and radiation therapy.
Chemotherapy for metastatic disease includes doxorubicin/ifosfamide or gemcitabine/docetaxel doublets, among others. Novel agents sometimes used as first-line therapy for MLPS includes the marine-derived compounds trabectedin and eribulin. Trabectedin binds to DNA, causing cell cycle arrest and apoptosis, but also seems to have a direct interaction with the FUS::DDIT3 gene product such that normal lipoblast maturation can occur. Eribulin can be used with multiple liposarcoma subtypes and is considered a microtubule inhibitor. Other targeted therapies under investigation for MLPS include immunotherapy, tyrosine kinase inhibitors, HIV protease inhibitors, and other medications targeting the FUS::DDIT3 gene product.
The identification of efficacious targeted therapies makes correct identification of MLPS imperative. Common differential diagnostic considerations for MLPS include other liposarcoma subtypes, myxofibrosarcoma, low-grade fibromyxoid sarcoma, and others. Most often, the differential diagnosis with these entities can be reliably resolved based upon careful histologic evaluation and the clinical setting. Molecular and immunohistochemical stains can aid greatly in challenging cases.
Well-differentiated liposarcoma/dedifferentiated liposarcoma (WDLPS/DDLPS) is the most common subtype of liposarcoma and usually arises within the retroperitoneum. Microscopically, WDLPS are characterized by the presence of adipocytes of varying size with intervening fibrous stroma and usually mild nuclear pleomorphism. DDLPS shows similar findings but with varying amounts of a distinct and highly cellular spindle cell-rich portion with 5 or more mitoses per 10 high-power fields.
Cytogenetic studies often reveal ring and giant marker chromosomes. In addition, these tumors show amplification of chromosome 12q13-15, the region containing the MDM2 gene, which inhibits p53 and the cell cycle regulator CDK4. Based on cytogenetic and molecular evidence, most consider DDPLS to be a dedifferentiation from WDPLS, and both entities are positive for MDM2 and CDK4 immunostains. WDLPS do not metastasize, while DDPLS metastasize in 10%-15% of cases, but both show locoregional recurrences.
Pleomorphic liposarcoma is the least common subtype of liposarcoma and does not contain a known single genetic aberration that would be considered characteristic. Instead, complex genetic abnormalities are seen, including multiple gene rearrangements, deletions, and gains. These complex genetic abnormalities result in the microscopic appearance of a high-grade sarcoma with high cellularity and pleomorphic nuclei, including pleomorphic and multinucleated lipoblasts. Adipocytic differentiation may be difficult to recognize, and in the past, these tumors were often called malignant fibrous histiocytoma. They follow a more aggressive clinical course than the other liposarcoma subtypes and do not respond well to current treatment modalities.
Myxofibrosarcoma (MF) is the most common sarcoma of elderly patients, with a slight male predominance. MF are usually superficial, involving the subcutis and dermis, although deep/subfascial lesions can occur in a minority of cases. Similar to MLPS, the lower extremities (thigh) are the most common site of origin, followed by the upper extremities and limb girdles. Microscopically, MF often contains alternating hypo- and hyper-cellular myxoid areas with occasional pleomorphic cells. The blood vessels in MF are typically curvilinear, compared to the plexiform or “crow’s feet” blood vessel pattern in MLPS. Pseudolipoblasts (containing mucin rather than lipid) may be seen, and mitoses may be numerous. These tumors are usually S100 negative, and they show a complex, but indistinct, karyotype without diagnostic chromosomal abnormalities.
Low-grade fibromyxoid sarcomas (LGFMS) lies along a histologic and molecular continuum that includes hyalinizing spindle cell tumor with giant rosettes. LGFMS, similar to MLPS, affects young and middle-aged adults, most often between the third and fifth decades, and usually presents as large deep/subfascial masses affecting the extremities. As with MLPS, the thigh is the most common site of origin for LGFMS.
Histologically, alternating fibrous and myxoid areas are appreciated, with a whorled growth pattern. Groupings or “arcades” of small blood vessels with perivascular hyalinization are often noted. The tumor cells are bland, short spindle cells with ovoid nuclei and without significant nuclear pleomorphism. Mitoses and necrosis are usually scarce. Giant rosettes are present in approximately one-third of cases. Immunohistochemistry often shows CD34, SMA or desmin immunoreactivity, while S100 is negative in LGFMS. MUC4 expression is diagnostically sensitive and specific for LGFMS. Two distinctive translocations, t(7;16)(q34;p11) and t(11;16)(p11;p11), can be identified in most cases. LGFMS has significant potential for both local recurrence and distant metastasis, frequently after a prolonged disease-free interval.
Supplementary Questions
- Which of the following is the most significant prognostic indicator in myxoid liposarcoma?
- A mitotic index of greater than 5 mitoses per 10 high-power fields.
- Identification of a CDKN2A mutation.
- p53 protein overexpression by immunohistochemistry.
- The presence of a round cell component (>5%).
- The presence of coagulative necrosis.
- Which of the following is the most useful aid in the diagnosis of myxoid liposarcoma?
- An immunostain panel that includes CD34, desmin, and SMA.
- Identification of t(12;16)(q13.p11) by cytogenetics or molecular analysis.
- Immunostains for MDM2 and CDK4.
- The presence of MUC4 immunopositivity.
- The presence of ring or giant marker chromosomes.
- Which of the following statements is true of myxoid liposarcoma?
- Myxoid liposarcomas are often radiation resistant.
- Myxoid liposarcomas often display a curvilinear rather than a plexiform vascular pattern.
- Myxoid liposarcomas usually arise in the retroperitoneum.
- Myxoid liposarcomas usually metastasize to sites other than lung parenchyma.
- Targeted therapies do not exist for myxoid liposarcoma
References
- Fletcher C, Hogendoorn P, Mertens F, et al. WHO Classification of Tumours of Soft Tissue and Bone. 4th edition. Lyon, France: IARC Press; 2013.
- Fritchie KJ, Goldblum JR, Tubbs RR, et al. The expanded histologic spectrum of myxoid liposarcoma with an emphasis on newly described patterns: Implications for diagnosis on small biopsy specimens. Am J Clin Pathol. 2012;137(2):229–239.
- Hoffman A, Ghadimi MP, Demicco EG, et al. Localized and metastatic myxoid/round cell liposarcoma. Cancer. 2013;119(10):1868–1877.
- Hornick JL (Ed). Practical Soft Tissue Pathology: A Diagnostic Approach. Philadelphia, PA: Elsevier Saunders; 2013, 141-147.
- Moreau LC, Turcotte R, Ferguson P, et al. Myxoid/round cell liposarcoma (MRCLS) revisited: an analysis of 418 primarily managed cases. Ann Surg Oncol. 2012;(4): 1081-1088.
- Muratori F, Bettini L, Frenos F, et al. Myxoid Liposarcoma: Prognostic Factors and Metastatic Pattern in a Series of 148 Patients Treated at a Single Institution. Int J Surg Oncol. 2018; volume 2018:8928706.
- Tseng WW, Somaiah N, Lazar AJ, Lev DC, and Pollack RE. Novel Systemic Therapies in Advanced Liposarcoma: A review of Recent Clinical Trial Results. Cancers(Basel). 2013;5: 529-49.
Author
Robert A. Schwartz, MD, FCAP
Surgical Pathology Committee
Eastern Connecticut Health Network and Waterbury Hospital
Manchester, Connecticut
Answer Key
- The presence of a round cell component (>5%). (d)
- Identification of t(12;16)(q13.p11) by molecular analysis. (b)
- Myxoid liposarcomas usually metastasize to sites other than lung parenchyma. (d)