- Home
- Member Resources
- Articles
- What’s New in Myelodysplastic Syndromes in Adults: ICC vs WHO5 and beyond

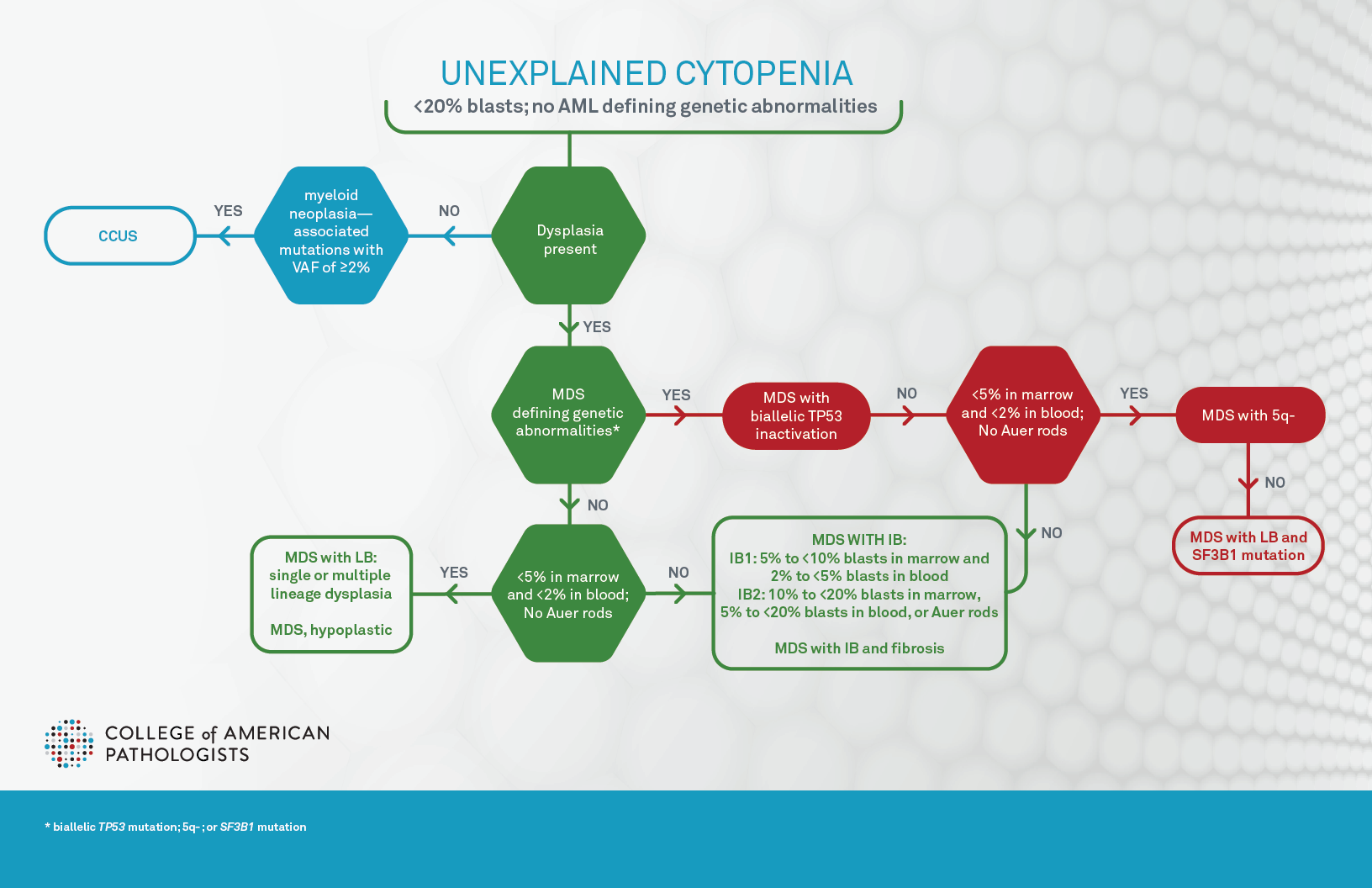
Diagnosis and classification of myelodysplastic syndromes (MDS) have significantly changed since 2022 with the advent of World Health Organization (WHO) 5th edition and International Consensus Classification (ICC) of hematopoietic neoplasms (both published in journal article form in 2022 and final book form in 2024 and 2025, respectively), subsequently referred to as WHO5 and ICC.1,2,3,4 Of note, WHO5 proposed to use “myelodysplastic neoplasm” to replace “myelodysplastic syndrome,” indicating the neoplastic nature of these diseases; however, it favored the abbreviation “MDS” over “MDN” to avoid the confusion between MDN and MPN. The goal of this article is to summarize the changes from the previous 2017 WHO classification revised 4th edition (aka WHO4r), and to highlight the differences between these new classifications (Figure 1A, Figure 1B, and Table 1). Additionally, molecular biomarker based prognostic stratification,5 recently proposed molecular classification,6 and the impact on pathology practice, particularly in terms of molecular hematopathology, will be briefly reviewed.

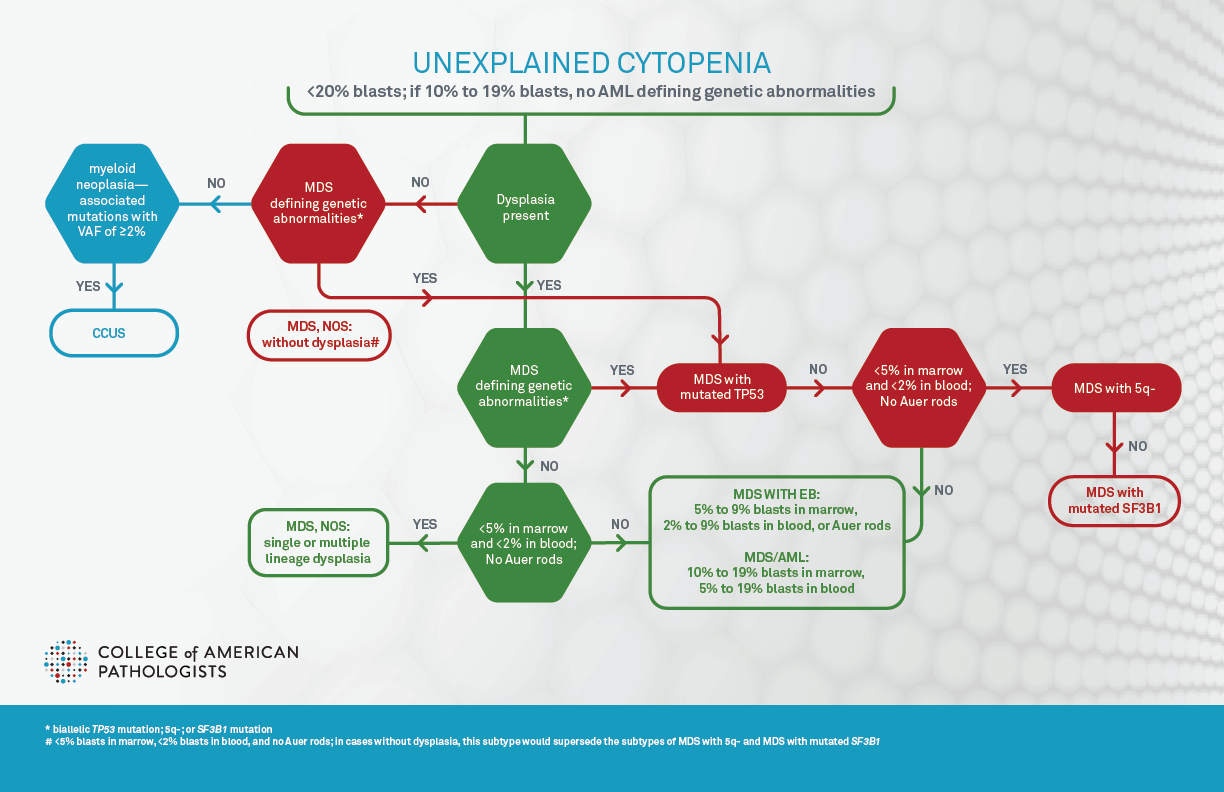
Both WHO5 and ICC define clonal cytopenia of undetermined significance (CCUS) as the precursor lesion of MDS. CCUS is clonal hematopoiesis (CH) detected in the presence of one or more persistent cytopenias, defined as anemia (hemoglobin <13 g/dL in males, <12 g/dL in females), neutropenia (neutrophil count <1.8 × 109/L), and/or thrombocytopenia (platelet count <150 × 109/L). Further discussion of CCUS can be found in another article on the CAP precision medicine page. The cytopenias are unexplained by reactive conditions and usually have persisted for four months or longer. Dysplastic changes can be seen, but must fall short of the diagnostic criteria for MDS (ie, <10% of cells showing dysplasia in each lineage), and blasts should not be increased. CH is defined as harboring chromosomal alterations, and/or one or more myeloid neoplasia–associated somatic driver mutations with variant allele frequency (VAF) of ≥2% or ≥4% for X-linked gene mutations in male patients. Of note, one critical discrepancy between ICC and WHO5 is that in ICC, patients with MDS-defining genetic abnormalities (Table 1 and Figure 1B) are excluded from CCUS and are instead classified as MDS with subtypes discussed below, since ICC allows absence of dysplasia in these patients for a diagnosis of MDS.
Both classification systems define MDS as a hematologic neoplasm with the presence of at least one unexplained cytopenia (as defined above and usually persistent for more than four months), MDS defining genetic abnormalities, and/or morphologic dysplasia in maturing hematopoietic elements (with the exception of specific genetic lesions in ICC). Sustained neutrophilia, monocytosis, and/ or thrombocytosis not explained by reactive conditions at the time of initial diagnosis generally warrant consideration as MDS/myeloproliferative neoplasm (MDS/MPN) or MPN. However, thrombocytosis (platelet count ≥450 × 109/L) is allowed in MDS-5q (see below). Persistent unexplained eosinophilia (eosinophils ≥10% of WBC and ≥1.5 × 109/L) would suggest an eosinophilic myeloid proliferation.
Both WHO5 and ICC acknowledge that dysplasia in one or more lineages is the morphological hallmark of MDS, with the threshold for dysplasia being 10% of cells for all lineages (ICC suggesting 30% or 40% may be more appropriate for megakaryocytic dysplasia), and that the blast percentage is <20%. Of note, cases with acute myeloid leukemia (AML) defining/recurrent genetic abnormalities should be classified as acute myeloid leukemia regardless of blast percentage by WHO5, or with ≥10% blasts by ICC with some exceptions (for example, cases with CEBPA mutation still require ≥20% blasts by WHO5, refer to What’s new in AML Classification (WHO 2022 vs International Consensus Classification)). Importantly, WHO5 requires the presence of dysplasia for the diagnosis of any subtypes of MDS, while ICC allows the diagnosis of four subtypes even in the absence of dysplasia: MDS with mutated TP53, MDS with del(5q), MDS with mutated SF3B1, and MDS, NOS without dysplasia (Table 1, Figure 1B). Both emphasize the importance of Auer rods, which, if present, leads to diagnosis of MDS with increased blasts-2 (MDS-IB2) in WHO5, or MDS with excess blasts in ICC regardless of blast percentage (Table 1). ICC further suggests that the presence of 1% peripheral blood blasts confirmed on two separate occasions is considered as MDS with excess blasts.
The most notable change in the new classifications is the introduction of genetically defining MDS subtypes (Table1). Both systems have introduced a new subtype based on multi-hit/biallelic TP53 mutations (MDS with mutated TP53 in ICC, and MDS with bi-allelic inactivation of TP53 in WHO5), to recognize the very poor prognosis for patients affected by these alterations. In WHO5, this subtype supersedes all subtypes of MDS, and in ICC this subtype supersedes all subtypes except MDS/AML subtype. However, there are differences regarding genetic profiles, variant allele frequency (VAF), and blast percentages. ICC defines this subtype as two distinct TP53 mutations, each with a VAF of ≥10%, or a single TP53 mutation with (1) 17p deletion, (2) VAF >50%, or (3) loss of heterozygosity (LOH) at the 17p TP53 locus. If TP53 locus LOH information is not available, complex karyotype with a single TP53 mutation is also accepted. In the WHO5 system, this subtype is defined by: two or more TP53 mutations (no VAF specified), one mutation with evidence of TP53 deletion or LOH, or one mutation with VAF >49% (presumptive of LOH). The blast threshold for this entity is less than 20% by WHO5 and less than 10% by ICC. In ICC, cases with ≥10% blasts are classified as MDS/AML with mutated TP53, which requires one mutated allele with variant allele frequency ≥10%.
The remaining genetically defined subtypes require that blasts are <5% in marrow and <2% in blood. The second subtype with a defining genetic abnormality is MDS with low blasts and isolated 5q deletion (MDS-5q) by WHO5 and MDS with del(5q) by ICC. This subtype is very similar to MDS with isolated del(5q) in WHO4r. Both classifications allow the del(5q) with up to one other abnormality, provided that the abnormality is not monosomy 7 or 7q deletion. The third subtype with a defining genetic abnormality is MDS with low blasts and SF3B1 mutation (VAF≥5%) by WHO5 and MDS with mutated SF3B1 (VAF ≥10%) by ICC, regardless of percentages of ringed sideroblasts. This is because recent research has shown that percentages of ring sideroblasts are not associated with favorable prognosis independent of the SF3B1 mutation. ICC uses this subtype to completely replace the MDS with ring sideroblasts subtype in the prior WHO4r, while WHO5 considers MDS with low blasts and ringed sideroblasts ≥15% as an acceptable alternative of MDS with low blasts and SF3B1 mutations for cases when SF3B1 is not tested in limited-resource settings and to include rare MDS cases with driver mutations in other RNA splicing components. Both WHO5 and ICC diagnoses preclude cases with isolated 5q-, −7/del(7q), or complex karyotype (defined as ≥3 cytogenetic abnormalities, excluding -Y). ICC further excludes cases with abnormal 3q26 and RUNX1 mutation. One subtype exists only in the ICC system, which is MDS, NOS without dysplasia, defined by cytogenetic abnormalities of -7, -7q, and complex karyotype.
The remaining processes are defined by morphologic dysplasia in ≥10% of cells in at least one lineage and blast percentages. When blasts are <5% in marrow and <2% in blood, ICC classifies this as MDS, NOS which is further classified into single or multilineage dysplasia, similar to WHO4r. In contrast, WHO5 classifies these cases as MDS with low blasts with a similar separation of single linage from multilineage dysplasia. WHO5 further adds a new subtype of hypoplastic MDS defined by <30% of normal cellularity in patients younger than 70 years and <20% in patients aged ≥70 years. Somatic mutation of the X-linked gene PIGA and a genetic predisposition to bone marrow failure should be excluded. This subtype is to recognize certain patients that may respond to immunosuppressive therapy.
Per WHO5, patients with ≥5% blasts in marrow, ≥2% in blood, and/or presence of Auer rods in blasts are classified as MDS with increased blasts grade 1 (5% to <10% blasts BM and/or 2% to <5% PB) and 2 (10% to <20% blasts BM and/or 5% to <20% PB; or Auer rods with blasts <20%), similar to WHO4r. Additionally, a new subtype MDS with increased fibrosis is added to recognize the fibrosis associated adverse prognosis. ICC classifies these scenarios as MDS with excess blasts (5%–9% blasts BM or 2%–9% PB; or Auer rods with blasts <10%) or MDS/AML (10%–19% blasts in BM and /or PB). The latter is then further subclassified using the AML classification schemes (please refer to What’s new in AML Classification (WHO 2022 vs International Consensus Classification)). This is to support participation in both MDS and AML clinical trials depending on clinical conditions. Of note, in accord with ICC, WHO5 also suggests that MDS-IB2 may be regarded as AML-equivalent for therapeutic considerations and from a clinical trial design perspective when appropriate.
In WHO5, MDS that arises secondary to exposure to cytotoxic therapy or germline predisposition are classified into the major category of myeloid neoplasms, secondary, with three sub-categories: myeloid neoplasm post cytotoxic therapy, myeloid neoplasms with associated germline predisposition, and myeloid proliferation associated with Down syndrome. In ICC, these conditions are considered as diagnostic qualifiers rather than classifiers. For example, in the hypothetical case of a patient with a history of soft tissue sarcoma post-chemotherapy who developed MDS with 8% blasts, TP53 mutation (VAF of 60%), and complex karyotype, the ICC diagnosis would be MDS with mutated TP53, therapy-related, while the WHO5 diagnosis would be myeloid neoplasm (MDS with biallelic TP53 inactivation), post cytotoxic therapy (MN-pCT).
Along with the new classification scheme of MDS, a clinical-molecular prognostic model (IPSS-Molecular [IPSS-M]) incorporating clinical and molecular variables has been developed to estimate leukemia-free survival, leukemic transformation probability, and overall survival in MDS patients.5 This is a significant milestone in MDS prognostication since risk stratification for MDS were traditionally based on only hematologic parameters and cytogenetic abnormalities, such as International Prognostic Scoring System–Revised (IPSS-R). IPSS-M uses four categories of features: cell counts (hemoglobin level, platelet count, and marrow blast percentage); IPSS-R cytogenetic category; 17 binary features from 16 prognostic genes (ASXL1, CBL, DNMT3A, ETV6, EZH2, FLT3, IDH2, KRAS, MLLPTD, NPM1, NRAS, RUNX1, SF3B15q, SF3B1α, SRSF2, TP53multihit, and U2AF1); and a feature representing the number of mutated genes from a residual group of 15 genes (BCOR, BCORL1, CEBPA, ETNK1, GATA2, GNB1, IDH1, NF1, PHF6, PPM1D, PRPF8, PTPN11, SETBP1, STAG2, and WT1). Of note, SF3B15q refers to concomitant presence with isolated del(5q), while SF3B1α refers to absence of co-mutation of SF3B1 and any of the genes BCOR, BCORL1, NRAS, RUNX1, SRSF2, or STAG2. Using different weight ratios for these features, IPSS-M then generates a unique risk score (which can be calculated via IPSS-M Risk Calculator) for patients to be classified into six IPSS-M risk groups with prognostic differences: very low, low, moderate low, moderate high, high, and very high.
Compared with the IPSS-R, the IPSS-M shows significant improvement for estimating leukemia-free survival, leukemic transformation probability, and overall survival in MDS patients, especially lower-grade MDS. Application of the IPSS-M re-stratified 46% of patients into different prognostic groups. Additionally, the IPSS-M is applicable in primary and secondary/therapy-related MDS. This new model will facilitate designing effective treatments for patients with different risks. For example, an international panel on behalf of the European Society for Blood and Marrow Transplantation recently suggested all patients with higher-risk MDS (moderate high, high, and very high) by IPSS-M are potential candidates for immediate allogeneic stem cells transplantation.7
Most recently, a molecular taxonomy of MDS, which may serve as a basis for a mechanistic MDS classification using genomic profiling data, has been proposed.6 This study defines 16 MDS molecular groups, encompassing 86% of patients, using information from 21 genes, 6 cytogenetic events, and LOH at the TP53 and TET2 loci. These groups are associated with distinct clinical phenotypes and outcomes. Although further studies are needed, the molecular based classification and prognostication of MDS appears very promising.
With the above changes, appropriate diagnosis, classification, and risk stratification of MDS cases will require integration of morphology, clinical features and study results of cytogenetics, FISH and/or next generation sequencing (NGS) targeting the genetic abnormalities. These latter methodologies are particularly important for application of the ICC in which dysplasia is not required in genetically defined subgroups. Consequently, it is likely that the volume of NGS testing, which has the potential to detect all these genetic abnormalities in a single assay (including fusions, copy number alterations, and copy neutral loss of heterozygosity), will significantly increase. The pathology communities should be aware of these changes and prepare to handle the increased testing volumes and keep up with the evolving classification and prognostication based on genetic abnormalities.
References
- D Arber, A Orazi, RP Hasserjian, et al. International Consensus Classification of Myeloid Neoplasms and Acute Leukemias: integrating morphologic, clinical, and genomic data, Blood 2022;140:1200.
- JD Khoury, E Solary, O Abla, et al The 5th edition of the World Health Organization Classification of Hematolymphoid Tumors: Myeloid and Histiocytic/Dendritic Neoplasms, Leukemia 2022;36:1703.
- WHO Classification of Tumours Editorial Board. Haematolymphoid tumours. Lyon (France): International Agency for Research on Cancer; 2024. (WHO classification of tumours series, 5th ed.; vol. 11). https://publications.iarc.who.int/637.
- Arber DA, Borowitz MJ, Cook JR, Leval L de, Goodlad JR, Hasserjian RP etal. (eds). The international consensus classification of myeloid and lymphoid neoplasms. Wolters Kluwer, 202
- Bernard E, Tuechler H, Greenberg PL, et al. Molecular International Prognostic Scoring System for Myelodysplastic Syndromes. NEJM Evid 2022; 1 (7).
- Bernard E, Hasserjian RP, Greenberg PL, Arango Ossa JE, Creignou M, Tuechler H et al. Molecular taxonomy of myelodysplastic syndromes and its clinical implications. Blood 2024; 144: 1617–1632.
- Carmelo Gurnari, et al. Clinical-genomic profiling of MDS to inform allo-HCT: recommendations from an international panel on behalf of the EBMT, Blood 2025:145:1987.

Chung-Che (Jeff) Chang, MD, PhD, FCAP, is the medical director of Hematology and Molecular/Genomic Laboratory at AdventHealth-Orlando, and professor of pathology, University of Central Florida. He currently serves as associate editor for Archives of Pathology and Laboratory Medicine, and a member of the Personalized Healthcare Committee for the College of American Pathologists (CAP). He was the principal investigator of several National Institutes of Health (NIH)/National Cancer Institute (NCI) grants to study myeloma, myelodysplastic syndromes, and lymphoma. Dr. Chang’s research interests include the use of next-generation sequencing technologies for clinical diagnostics and biomarker discovery.